 PDF
PDF  Views
Views  Share
Share


Lower Intestinal Langerhans Cell Histiocytosis Masquerading as Chronic Malabsorption Syndrome and Failure to Thrive in a Child: A Rare Case Presented with a Succinct Review of Recent Literature
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2018; 39(04): 543-545
DOI: DOI: 10.4103/ijmpo.ijmpo_73_17
Abstract
Langerhans cell histiocytosis (LCH) encompasses a group of disorders characterized by neoplastic proliferation of Langerhans cells causing destruction of tissue, morbidity and mortality. The authors bring forth the case of a 2-year-old child presenting with chronic diarrhea and features of chronic malabsorption that revealed lesions over the ileocolonic mucosa. Histopathological examination with immunohistochemical expression for CD1a confirmed the diagnosis of LCH. Gastrointestinal LCH is quite uncommon, and review of the published English literature revealed about 50 reported cases. The importance of diagnosing this disease is echoed by the grim survival rate of 18 months from the time of diagnosis that is seen in around 60% of cases. Owing to its rarity and unfamiliarity of practicing physicians and pathologists of this disease at this site, the authors wish to discuss the potential pitfalls and differential diagnoses.
Publication History
Article published online:
17 June 2021
© 2018. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Langerhans cell histiocytosis (LCH) encompasses a group of disorders characterized by neoplastic proliferation of Langerhans cells causing destruction of tissue, morbidity and mortality. The authors bring forth the case of a 2-year-old child presenting with chronic diarrhea and features of chronic malabsorption that revealed lesions over the ileocolonic mucosa. Histopathological examination with immunohistochemical expression for CD1a confirmed the diagnosis of LCH. Gastrointestinal LCH is quite uncommon, and review of the published English literature revealed about 50 reported cases. The importance of diagnosing this disease is echoed by the grim survival rate of 18 months from the time of diagnosis that is seen in around 60% of cases. Owing to its rarity and unfamiliarity of practicing physicians and pathologists of this disease at this site, the authors wish to discuss the potential pitfalls and differential diagnoses.
Introduction
Langerhans cell histiocytosis (LCH) is a clonal proliferation of histiocyte-like cells with a unique spectrum of tissue involvement that can range from self-limited disease to a disseminated highly explosive and life-threatening disease.[1] Children are the frequently affected with the disease having an affinity toward the head and neck region and osteoarticular system. While a wide plethora of organ systems may be affected and no particular age group is exempt from this disease process, involvement of the gastrointestinal (GI) tract is exceedingly rare.[2]
Case Report
A 2-year-old girl child belonging to one of our neighboring countries presented to the gastroenterology clinic with chief complaints of recurrent diarrhea for 3 months of age and recent onset of serous otorrhea. Her parents complained of malabsorption, watery diarrhea, and failure to gain weight. General physical examination showed features of dehydration and marasmus with mild hepatosplenomegaly and a few purpuric macules over the abdomen. Upper GI and lower GI endoscopies were planned as routine malabsorption workup, and a subsequent dermatology consult was also sought. Upper GI endoscopy showed gastric angioectasia whereas the colonoscopy showed scattered erythematous spots over the visualized terminal ileum and colorectal mucosa. Mucosal biopsies showed mild architectural distortion with expansion of the lamina propria by clusters and nests of polygonal cells displaying round to reniform nuclei with open chromatin, few with visible nuclear grooves, distinct nucleoli, and an appreciable amount of eosinophilic cytoplasm [Figure 1]. Histology was suspicious and immunohistochemical (IHC) makers were ordered. The cells showed strong positivity for S-100 and CD1a [Figure 2] which confirmed the diagnosis of LCH involving the lower intestinal tract. Subsequent biopsies from skin lesions also revealed LCH. Complete body computed tomography imaging showed lytic bone and liver lesions. The patient was managed with aggressive chemotherapy in view of multicentric disease and initiated with a combination of prednisolone and vinblastine for 6 weeks. The patient’s parents were advised about the poor prognosis, and they wished to take the treatment protocol and continued treatment in their own country. Within 5 months after initial diagnosis, the child had succumbed to her disease.
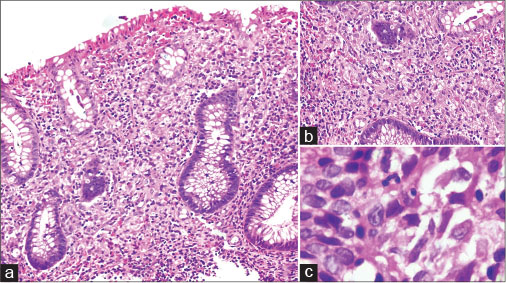
| Figure 1:(a and b) Colonic mucosal biopsy shows expansion of lamina propria by polygonal cells with mixed inflammatory infiltrate and conspicuous eosinophils (H and E, ×100 and ×200). (c) High power shows vesicular nuclei, intranuclear grooving and moderate amounts of eosinophilic cytoplasm and indistinct cell membranes (H and E, ×400)
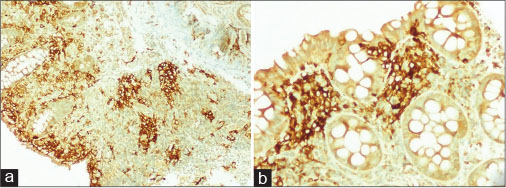
| Figure.2:Immunohistochemical staining with CD1a antibody shows strong reactivity for neoplastic cells within the lamina propria (×40 and ×200)
Discussion
LCH is a neoplasm of histiocytes that are derived from a common progenitor cell in the hematopoietic marrow.[1] [2] [3] [4] These histiocytes bear resemblance to the Langerhans cells that normally reside within the mucosa and skin, presenting antigens to our immune system for further processing. The etiology remains obscure and controversial, leading some to argue that LCH may be a reactive process rather than a neoplastic proliferation. However, recent literature has proved LCH to be a clonal proliferation with recurrent activation mutations of BRAF V600E in up to 60% of patients.[1] [5] Some authors believe that an “inflammatory myeloid neoplasia” be a more acceptable terminology until further research sheds light into the exact pathogenesis while some advocate that LCH be considered a “myeloproliferative neoplasm” since mutations that are associated with the pathogenesis of LCH are not uncommon in the tumors that arise from hematopoietic stem cells and myeloid precursors.[1]
Historically, LCH was subclassified as “eosinophilic granuloma” if the disease was limited to a particular site only; “Hand-Schuller-Christian disease” for unifocal and multisystemic disease; and as “Letterer–Siwe disease” in case of multifocal and multisystemic disease.[1] [5] [6] An underreported self-healing form referred to as “Hashimoto-Pritzker disease” is also described. In 1953, Lichtenstein unified these clinical entities under the term “Histiocytosis-X” later to be changed into the current terminology of LCH, in 1983.[6] LCH has been found to occur in any age group, but it is usually seen in the pediatric age group with a peak in its incidence for children below 2 with males being twice as common and females.[1] Its reported incidence is around 4–8/million children and 1–2/million adults.[1] Unfortunately, no organ system is exempt although LCH most commonly occurs in the skin and osteoarticular system.[3] [7] [8]
LCH involving the GI tract is exceedingly rare,[4] [9] [10] [11] existing in the English literature as published case reports and occasional series, the largest one comprising of 12 cases.[2] To the best of our knowledge and after an extensive literature search through PubMed, around 53 reported cases of intestinal LCH exist in the English literature.[2] [4] [9] [12] Accordingly, our present case would represent possibly the 37th pediatric case of intestinal LCH in the English medical literature.
GI tract LCH is mostly seen in boys younger than 24 months of age with multisystemic disease at the time of presentation.[1] Nearly all reported children have been symptomatic presenting with nonspecific complaints such as nausea, vomiting, abdominal discomfort, diarrhea, failure to thrive, and malabsorption.[2] [4] Protein losing enteropathy and intestinal perforation have also occurred. In striking contrast to children are the adult cases, where more than 50% of patients are asymptomatic and have a female predominance. Endoscopically, lesions in pediatric age group are multiple, erythematous, and ulcerative whereas in the adults population, they are solitary and polypoidal projections in the mucosa.[2] [11] [12] Childhood intestinal LCH has a poor prognosis as no specific chemotherapeutic strategy has shown promise. More than 50% of children die within 18 months from the point of diagnosis.[2] [4] [10] [12]
Histopathological features are similar among the described GI tract lesions. The lamina propria shows patchy clusters, nests, or sheets of histiocytic cells with kidney-bean-shaped nuclei, irregular nuclear contours, and intranuclear grooves. Nuclear chromatin is finely dispersed and in some cases shows a prominent nucleolus. The inflammatory background has a conspicuous component of eosinophils. On immunohistochemistry, the cells display strong immunopositivity for S-100 protein, CD1a, and Langerin.[2] [4] Tennis racquet-shaped “Birbeck-granules” are seen on ultrastructural studies.
Differential diagnosis can include Crohn’s disease, Juvenile xanthogranuloma (JXG), and other rare histiocytic proliferations such as Rosai–Dorfman disease (RDD) and rarely histiocytic sarcoma. Careful histological assessment with IHC evidence will ultimately aid in the differentiation.
The absence of Touton-like giant cells and negativity for CD1a will exclude JXG. Extranodal RDD shows emperipolesis, a plasma cell-rich background and is nonreactive for CD1a or Langerin. Histiocytic sarcoma shows anaplasia with numerous CD68-positive and CD1a-negative histiocytes. Since malignant melanoma is the most common metastatic malignancy to the GI tract, it should also be excluded, especially in the adult cases. Melanoma cells are S-100 positive, HMB-45 and Melan-A positive, but CD1a is consistently negative.
Conclusion
Intestinal LCH is an exceedingly rare disease that has a predilection for the pediatric age group and unlike the adult cases has a grim prognosis owing to its widespread and multisystemic involvement at the time of presentation and its poor response to chemotherapy. Due to the extreme uncommon nature of LCH to occur in the GI tract, pathologists might not be familiar and entertain this entity as a differential diagnosis. Prompt diagnosis with ICH evidence is essential for aggressive patent management and early initiation of treatment.
Conflict of Interest
There are no conflicts of interest.
References
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood 2015; 126: 26-35
- Singhi AD, Montgomery EA. Gastrointestinal tract langerhans cell histiocytosis: A clinicopathologic study of 12 patients. Am J Surg Pathol 2011; 35: 305-10
- A multicentre retrospective survey of Langerhans’ cell histiocytosis: 348 cases observed between 1983 and 1993. The French Langerhans’ Cell Histiocytosis Study Group. Arch Dis Child 1996; 75: 17-24
- Geissmann F, Thomas C, Emile JF, Micheau M, Canioni D, Cerf-Bensussan N. et al. Digestive tract involvement in Langerhans cell histiocytosis. The French Langerhans Cell Histiocytosis Study Group. J Pediatr 1996; 129: 836-45
- Grana N. Langerhans cell histiocytosis. Cancer Control 2014; 21: 328-34
- Howarth DM, Gilchrist GS, Mullan BP, Wiseman GA, Edmonson JH, Schomberg PJ. Langerhans cell histiocytosis: Diagnosis, natural history, management, and outcome. Cancer 1999; 85: 2278-90
- Favara BE, Feller AC, Pauli M, Jaffe ES, Weiss LM, Arico M. et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol 1997; 29: 157-66
- Risdall RJ, Dehner LP, Duray P, Kobrinsky N, Robison L, Nesbit Jr. ME. Histiocytosis X (Langerhans’ cell histiocytosis). Prognostic role of histopathology. Arch Pathol Lab Med 1983; 107: 59-63
- Sharma S, Gupta M. A colonic polyp due to Langerhans cell histiocytosis: A lesion not to be confused with metastatic malignant melanoma. Histopathology 2006; 49: 438-9
- Shankar U, Prasad M, Chaurasia OP. A rare case of langerhans cell histiocytosis of the gastrointestinal tract. World J Gastroenterol 2012; 18: 1410-3
- Abdullgaffar B, Al-Murbati B, Al-Falasi M, Al-Otaibi L. Unsuspected Langerhans cell histiocytosis can be easily missed in a colonic biopsy. Fetal Pediatr Pathol 2014; 33: 98-103
- Shima H, Takahashi T, Shimada H. Protein-losing enteropathy caused by gastrointestinal tract-involved Langerhans cell histiocytosis. Pediatrics 2010; 125: e426-32
Address for correspondence
Publication History
Article published online:
17 June 2021
© 2018. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2,
Noida-201301 UP, India
| Figure 1:(a and b) Colonic mucosal biopsy shows expansion of lamina propria by polygonal cells with mixed inflammatory infiltrate and conspicuous eosinophils (H and E, ×100 and ×200). (c) High power shows vesicular nuclei, intranuclear grooving and moderate amounts of eosinophilic cytoplasm and indistinct cell membranes (H and E, ×400)
| Figure.2:Immunohistochemical staining with CD1a antibody shows strong reactivity for neoplastic cells within the lamina propria (×40 and ×200)
References
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood 2015; 126: 26-35
- Singhi AD, Montgomery EA. Gastrointestinal tract langerhans cell histiocytosis: A clinicopathologic study of 12 patients. Am J Surg Pathol 2011; 35: 305-10
- A multicentre retrospective survey of Langerhans’ cell histiocytosis: 348 cases observed between 1983 and 1993. The French Langerhans’ Cell Histiocytosis Study Group. Arch Dis Child 1996; 75: 17-24
- Geissmann F, Thomas C, Emile JF, Micheau M, Canioni D, Cerf-Bensussan N. et al. Digestive tract involvement in Langerhans cell histiocytosis. The French Langerhans Cell Histiocytosis Study Group. J Pediatr 1996; 129: 836-45
- Grana N. Langerhans cell histiocytosis. Cancer Control 2014; 21: 328-34
- Howarth DM, Gilchrist GS, Mullan BP, Wiseman GA, Edmonson JH, Schomberg PJ. Langerhans cell histiocytosis: Diagnosis, natural history, management, and outcome. Cancer 1999; 85: 2278-90
- Favara BE, Feller AC, Pauli M, Jaffe ES, Weiss LM, Arico M. et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol 1997; 29: 157-66
- Risdall RJ, Dehner LP, Duray P, Kobrinsky N, Robison L, Nesbit Jr. ME. Histiocytosis X (Langerhans’ cell histiocytosis). Prognostic role of histopathology. Arch Pathol Lab Med 1983; 107: 59-63
- Sharma S, Gupta M. A colonic polyp due to Langerhans cell histiocytosis: A lesion not to be confused with metastatic malignant melanoma. Histopathology 2006; 49: 438-9
- Shankar U, Prasad M, Chaurasia OP. A rare case of langerhans cell histiocytosis of the gastrointestinal tract. World J Gastroenterol 2012; 18: 1410-3
- Abdullgaffar B, Al-Murbati B, Al-Falasi M, Al-Otaibi L. Unsuspected Langerhans cell histiocytosis can be easily missed in a colonic biopsy. Fetal Pediatr Pathol 2014; 33: 98-103
- Shima H, Takahashi T, Shimada H. Protein-losing enteropathy caused by gastrointestinal tract-involved Langerhans cell histiocytosis. Pediatrics 2010; 125: e426-32