 PDF
PDF  Views
Views  Share
Share


Multiple Inherited Schwannomas, Meningiomas, and Ependymomas Syndrome in an Adult Patient
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2020; 41(02): 266-268
DOI: DOI: 10.4103/ijmpo.ijmpo_141_18
Abstract
Neurofibromatosis type 2 (NF2) is also known as multiple inherited schwannomas, meningiomas, and ependymomas (MISME) syndrome. Mutation in NF2 gene is the cause for MISME syndrome. We are reporting here a case of MISME syndrome with triple tumor in a 30-year-old male patient who presented with the chief complaints of spastic paraparesis, bowel and bladder incontinence, and decreased vision in the right eye.
Publication History
Received: 25 June 2018
Accepted: 12 September 2018
Article published online:
23 May 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Neurofibromatosis type 2 (NF2) is also known as multiple inherited schwannomas, meningiomas, and ependymomas (MISME) syndrome. Mutation in NF2 gene is the cause for MISME syndrome. We are reporting here a case of MISME syndrome with triple tumor in a 30-year-old male patient who presented with the chief complaints of spastic paraparesis, bowel and bladder incontinence, and decreased vision in the right eye.
Keywords
Ependymoma - meningioma - multiple inherited schwannomas - and neurofibromatosisIntroduction
Neurofibromatosis type 2 (NF2) incidence is 1 in 33,000 and prevalence is 1 in 60,000.[1] [2] It has no predilection for sex, race, and ethnicity, and it is most commonly seen in the second and third decades of life that too most commonly between 16 and 24 years of age.[3] Approximately 50% of cases are familial and remaining 50% are sporadic in nature.[4] NF2 is caused by the mutation in merlin gene, which is located on the long arm of chromosome 22 (22q12.2).[5] The hallmark for the diagnosis of NF2 is bilateral vestibular schwannomas on magnetic resonance imaging (MRI) of the brain. Here, we are reporting a rare case of multiple inherited schwannomas, meningiomas, and ependymomas (MISME) syndrome or triple tumor in a 30-year-old patient presenting with unilateral vestibular schwannomas, frontal meningioma, and histopathologically confirmed intramedullary ependymoma at L3-L5 region and intramedullary schwannomas at D12-L2.
Case Report
A thirty-year-old male patient presented to our hospital outpatient department in December 2014 with the chief complaints of lower back pain for the past 1 year weakness in bilateral lower limb for the past 1 year, urinary incontinence for the past 15 days and decreased vision in the left eye for the past 1 week. Neurological examination of motor system shows tone to be normal in the upper limb and power grade 5/5 in the bilateral upper limb. Tone reduced in bilateral lower limbs. Deep tendon reflex was absent in bilateral lower limbs. Pure-tone audiometry showed bilateral sensory neural deafness, which was more on the right side. Ophthalmic evaluation was done and was suggestive of macular corneal opacity of the left eye and central serous retinopathy with nystagmus in the right eye. MRI brain with contrast showed a well-defined enhancing extra-axial space-occupying lesion (SOL) at left cerebellopontine (CP) angle largest measuring 1.4 cm × 1.2 cm. The lesion is showing typical ice-cream cone appearance of CP angle mass with a smaller extracanalicular component. There was a distinct cerebrospinal fluid cleft between SOL and adjacent cerebellum, which was suggestive of acoustic schwannoma [Figure 1]. Multiple well-defined round-to-oval extra-axial-based homogeneously enhancing soft-tissue lesions largest measuring 3.2 cm × 2.2 cm × 2.0 cm were seen in the bilateral frontal, right temporal, right parietal region, right cerebellar hemisphere, and right CP angle and along the falx in midline which is suggestive of meningioma [Figure 2]a, [Figure 2]b, [Figure 2]c. Perifocal edema and dural thickening were also noted around few of the lesions. Contrast-enhanced MRI of the lumbosacral spine with screening of dorsal and cervical spine study showed a focal well-defined intradural SOL of size approximately 8.1 cm × 1.5 cm × 1.6 cm seen at D12 to L2 vertebral levels related to the conus medullaris, nearly completely occupying the thecal sac with ill-defined outline of the conus medullaris [Figure 3]. Another small intradural enhancing lesion (10 mm in size) was seen at L3 vertebral level inferior to the above lesion and another large intradural lesion (5 cm × 1.6 cm × 1.2 cm) at L4 vertebral level. The above findings are suggestive of multiple intraspinal schwannomas large lesion at D12-L2 and L4-L5 vertebral levels [Figure 3]. The patient has undergone D11 partial D12-L5 laminectomy with excision of D12-L2 intramedullary SOL and L3-L5 multiple intradural SOL. Histopathological examination of two lesions at vertebral site L3-L5 SOL showed ependymoma [Figure 4], and D12-L2 SOL showed schwannoma [Figure 5].
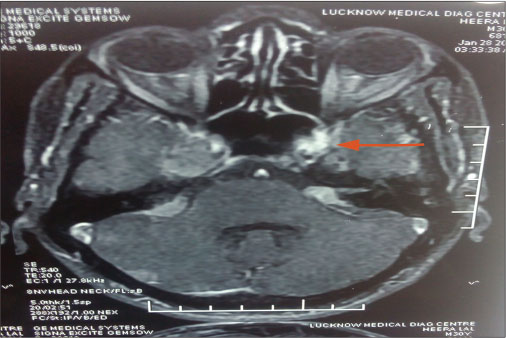
| Figure 1: A well-defined enhancing extra-axial space-occupying lesion at left cerebellopontine angle largest measuring 1.4 cm × 1.2 cm. The lesion is showing typical ice-cream cone appearance of cerebellopontine angle mass with a smaller extracanalicular component. There was a distinct cerebrospinal fluid cleft between space-occupying lesion and adjacent cerebellum, which was suggestive of acoustic schwannoma
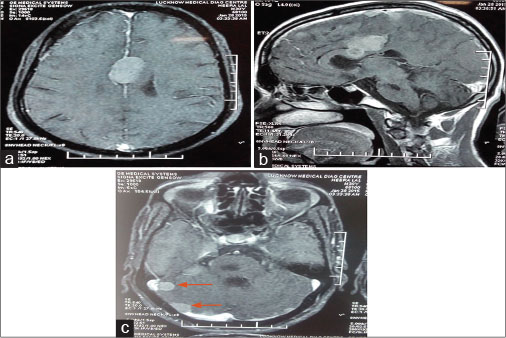
| Figure 2: (a–c) Multiple well-defined round-to-oval extra-axial-based homogenously enhancing soft-tissue lesions largest 3.2 cm × 2.2 cm × 2.0 cm were seen in the bilateral frontal, right temporal, right parietal region, right cerebellar hemisphere, and right cerebellopontine angle and along the falx in midline which is suggestive of meningioma
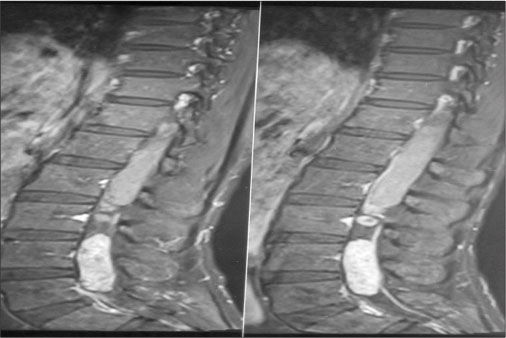
| Figure 3: (a–c) A focal well-defined intradural space-occupying lesion of size approximately 8.1 cm × 1.5 cm × 1.6 cm seen at D12 to L2 vertebral levels related to the conus medullaris, nearly completely occupying the thecal sac with an ill-defined outline of the conus medullaris
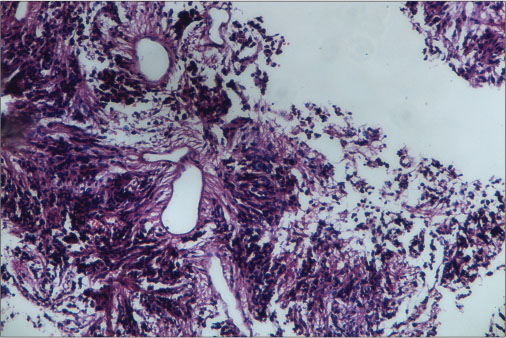
| Figure 4: Tumor arranged around blood vessels forming pseudorosettes as well as lying singly (H and E)
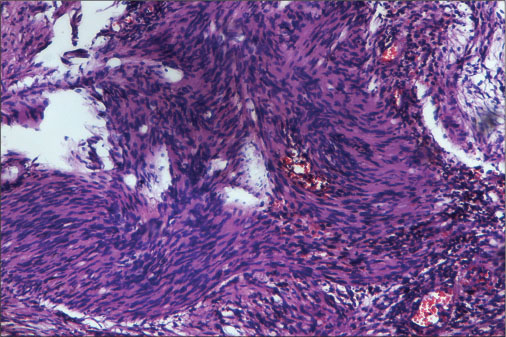
| Figure 5: Show predominantly hyperdense (Antoni A) with the hypodense area along with few congested blood vessels. Hyperdense areas (Antoni A) show palisade appearance (Verocay) of cells (H and E)
Discussion
NF2 is basically characterized by neoplasms in the central nervous system and peripheral nervous system. Neurofibromatosis was classified into two types based on their clinical and pathological features.[6] About 90%–95% of cases of NF2 have eighth cranial nerve schwannomas, and two-thirds of patients develop spinal neoplasms.[7] [8] Nearly 90% of patients suffer from ocular abnormalities with most of them presenting with early cataract, but retinal hamartomas and corneal lesions have also been documented. Definitive diagnosis of NF2 is bilateral eighth cranial nerve schwannomas on imaging or first-degree relative with NF2 and unilateral eighth cranial schwannoma at <30 years or any two of following: glioma, schwannoma, neurofibroma, meningioma, and juvenile posterior subcapsular lenticular opacity. The criteria for probable or presumptive diagnosis of NF2 are unilateral eighth cranial schwannoma before the age of 30 years and glioma, schwannoma, neurofibroma, meningioma or cortical cataract or posterior subcapsular cataract or unilateral vestibular schwannoma <30 years, and multiple meningiomas (two or more) or at least one schwannoma, glioma, and juvenile lens opacity.[9] [10] [11] [12] The investigation of choice in NF2 patients is the MRI of the brain and spine with contrast. NF2 patients should be managed by multidisciplinary team which includes neuroradiologist, neurologists, neurosurgeons, ophthalmologist, otologist, audiologist, and geneticist. Few cases have been reported with the occurrence of all three tumors, i.e., schwannomas, meningioma, and ependymoma in a single patient. In this case report, an adult patient with triple tumor unilateral acoustic schwannoma, right frontal meningioma, lumbar intramedullary ependymoma, and dorsal intramedullary schwannoma has been presented.
Conclusion
NF2 or MISME syndrome is rare in occurrence, in which the development of bilateral eighth cranial nerve schwannomas is must for diagnosis. Treatment is mainly focused on the maintenance of quality of life and preservation of cranial nerve and their function, as there is always a tendency of formation of new tumors. Simultaneous occurrence of triple tumors in an NF2 patient is rare. To rule out the possibility of tumors at a different location, we should get the imaging of the brain and whole spine while treating these cases.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Conflict of Interest
There are no conflicts of interest.
References
- Jin X, He J, Wang D, Zhang X, Wu Y, Liu H. et al. MISME syndrome with triple tumors affecting cervical spinal cord. Neurol Cases 2014; 1: 2374-3522
- Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM. et al. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am J Med Genet A 2010; 152A: 327-32
- Baser ME, Evans DG. Lack of sex-ratio distortion in neurofibromatosis 2. Am J Med Genet 2000; 95: 292
- Evans DG. Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J Rare Dis 2009; 4: 16
- Antinheimo J, Sankila R, Carpén O, Pukkala E, Sainio M, Jääskeläinen J. et al. Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology 2000; 54: 71-6
- Striedinger K, VandenBerg SR, Baia GS, McDermott MW, Gutmann DH, Lal A. et al. The neurofibromatosis 2 tumor suppressor gene product, merlin, regulates human meningioma cell growth by signaling through YAP. Neoplasia 2008; 10: 1204-12
- Chen AF, Samy RN, Gantz BJ. Cerebellopontine angle tumor composed of Schwann and meningeal proliferations. Arch Otolaryngol Head Neck Surg 2001; 127: 1385-9
- Asthagiri AR, Parry DM, Butman JA, Kim HJ, Tsilou ET, Zhuang Z. et al. Neurofibromatosis type 2. Lancet 2009; 373: 1974-86
- Walter J, Kuhn SA, Brodhun M, Reichart R, Kalff R. Pulmonary meningioma and neurinoma associated with multiple CNS tumours in a patient with neurofibromatosis type 2. Clin Neurol Neurosurg 2009; 111: 454-9
- Stumpf DA, Alksne JF, Annegers JF. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 1988; 45: 575-8
- Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE. et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997; 278: 51-7
- National Institutes of Health Consensus Development Conference Statement on Acoustic Neuroma, December 11-13 1991. The consensus development panel. Arch Neurol 1994; 51: 201-7
Address for correspondence
Publication History
Received: 25 June 2018
Accepted: 12 September 2018
Article published online:
23 May 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Figure 1: A well-defined enhancing extra-axial space-occupying lesion at left cerebellopontine angle largest measuring 1.4 cm × 1.2 cm. The lesion is showing typical ice-cream cone appearance of cerebellopontine angle mass with a smaller extracanalicular component. There was a distinct cerebrospinal fluid cleft between space-occupying lesion and adjacent cerebellum, which was suggestive of acoustic schwannoma
| Figure 2: (a–c) Multiple well-defined round-to-oval extra-axial-based homogenously enhancing soft-tissue lesions largest 3.2 cm × 2.2 cm × 2.0 cm were seen in the bilateral frontal, right temporal, right parietal region, right cerebellar hemisphere, and right cerebellopontine angle and along the falx in midline which is suggestive of meningioma
| Figure 3: (a–c) A focal well-defined intradural space-occupying lesion of size approximately 8.1 cm × 1.5 cm × 1.6 cm seen at D12 to L2 vertebral levels related to the conus medullaris, nearly completely occupying the thecal sac with an ill-defined outline of the conus medullaris
| Figure 4: Tumor arranged around blood vessels forming pseudorosettes as well as lying singly (H and E)
| Figure 5: Show predominantly hyperdense (Antoni A) with the hypodense area along with few congested blood vessels. Hyperdense areas (Antoni A) show palisade appearance (Verocay) of cells (H and E)
- 1 Jin X, He J, Wang D, Zhang X, Wu Y, Liu H. et al. MISME syndrome with triple tumors affecting cervical spinal cord. Neurol Cases 2014; 1: 2374-3522
- 2 Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM. et al. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am J Med Genet A 2010; 152A: 327-32
- 3 Baser ME, Evans DG. Lack of sex-ratio distortion in neurofibromatosis 2. Am J Med Genet 2000; 95: 292
- 4 Evans DG. Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J Rare Dis 2009; 4: 16
- 5 Antinheimo J, Sankila R, Carpén O, Pukkala E, Sainio M, Jääskeläinen J. et al. Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology 2000; 54: 71-6
- 6 Striedinger K, VandenBerg SR, Baia GS, McDermott MW, Gutmann DH, Lal A. et al. The neurofibromatosis 2 tumor suppressor gene product, merlin, regulates human meningioma cell growth by signaling through YAP. Neoplasia 2008; 10: 1204-12
- 7 Chen AF, Samy RN, Gantz BJ. Cerebellopontine angle tumor composed of Schwann and meningeal proliferations. Arch Otolaryngol Head Neck Surg 2001; 127: 1385-9
- 8 Asthagiri AR, Parry DM, Butman JA, Kim HJ, Tsilou ET, Zhuang Z. et al. Neurofibromatosis type 2. Lancet 2009; 373: 1974-86
- 9 Walter J, Kuhn SA, Brodhun M, Reichart R, Kalff R. Pulmonary meningioma and neurinoma associated with multiple CNS tumours in a patient with neurofibromatosis type 2. Clin Neurol Neurosurg 2009; 111: 454-9
- 10 Stumpf DA, Alksne JF, Annegers JF. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 1988; 45: 575-8
- 11 Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE. et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997; 278: 51-7
- 12 National Institutes of Health Consensus Development Conference Statement on Acoustic Neuroma, December 11-13 1991. The consensus development panel. Arch Neurol 1994; 51: 201-7