 PDF
PDF  Views
Views  Share
Share


Neonatal Kasabach-Merritt phenomenon
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2011; 32(04): 238-241
DOI: DOI: 10.4103/0971-5851.95150
Abstract
Kasabach-Merritt phenomenon (KMP) is a life-threatening consumptive coagulopathy in the presence of a rapidly enlarging vascular tumor. It usually presents in early infancy, but onset in early neonatal period, facial hemangioma, and vincristine use in neonates has rarely been reported. We, hereby, present a 6-day-old male child presenting with facial hemangioma and intracranial hemorrhage, and KMP responding well to steroids and vincristine. Pathophysiology of disease and various treatment options have been discussed.
Publication History
Article published online:
06 August 2021
© 2011. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Kasabach-Merritt phenomenon (KMP) is a life-threatening consumptive coagulopathy in the presence of a rapidly enlarging vascular tumor. It usually presents in early infancy, but onset in early neonatal period, facial hemangioma, and vincristine use in neonates has rarely been reported. We, hereby, present a 6-day-old male child presenting with facial hemangioma and intracranial hemorrhage, and KMP responding well to steroids and vincristine. Pathophysiology of disease and various treatment options have been discussed.
INTRODUCTION
Kasabach-Merritt phenomenon (KMP) is a life-threatening, consumptive coagulopathy associated with an underlying vascular tumor. It is characterized by severe thrombocytopenia, microangiopathic anemia, hypofibrinogenemia, and elevated fibrin split products in the presence of a rapidly enlarging tumor. KMP usually presents in early infancy and commonly reported sites of tumor include extremities, trunk, and neck.[1] There is no consensus in treatment and various regimens have been used by different authors. We, hereby, report a child with KMP, presenting in early neonatal period with facial hemangioma and responding to steroids and vincristine.
CASE REPORT
A 6-day-old male baby was brought to emergency with swelling over right half of face since birth and jaundice and lethargy for 2 days. Swelling was soft, erythematous 2 × 2 cm in size at birth over right cheek which increased progressively over next 6 days to involve right half of face extending from angle of mandible to scalp including pinna and right eye. Parents also noticed progressively increasing yellowish discoloration of skin and sclera for 2 days and lethargy and decreased oral acceptance for one day. There was no history of fever, trauma, bleeding from any site, seizures, ear or eye discharge. He was passing urine and stools normally. Baby was born at term gestation by normal vaginal delivery at home and cried immediately after birth. He was a good size baby, though exact birth weight was not recorded and was exclusively breastfed. There was no history of prolonged or difficult labor. He was the first child born of non consanguineous marriage and antenatal period was uneventful.
On examination, child was sick and lethargic with weight 2.420 kg and normal head circumference (35.5 cm). Child was hemodynamically stable with wide open (3 × 3 cm) and bulging anterior fontanelle. Large, indurated, erythematous, non-pulsatile swelling was present over whole of right half of the face, which was firm in consistency and local temperature was not elevated [Figure 1]. Child also had severe pallor and icterus till umbilicus. There was no external bleeding except right eye conjuctival hemorrhage. Child did not have any other gross congenital malformation. Central nervous system examination revealed lethargic child with depressed neonatal reflexes and upgoing plantars. Liver was palpable 3 cm below coastal margins (liver span 5.5 cm) and spleen was not palpable. Nasogastric tube aspiration did not reveal any gastric bleed. Cardiovascular and respiratory system examination was normal.
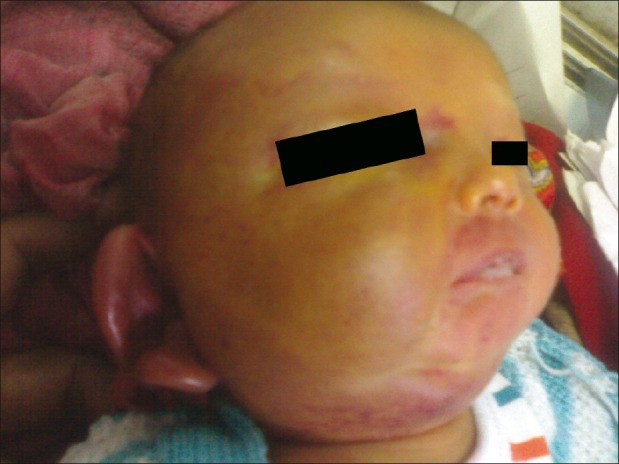
| Figure 1:At presentation. Hemangioma involving right half of face and earlobe
Initial blood investigations revealed severe anemia (Hb 5.4 gm/dl), normal total and differential leucocyte count and severe thrombocytopenia (platelet count 16,000/μL). Peripheral smear revealed features of hemolysis with severe thrombocytopenia. Neonatal sepsis screen was positive and coagulation profile was deranged with prolonged prothrombin (international normalized ratio [INR] 2.4), activated partial thromboplastin time (prolonged 32 seconds above control) and elevated D-dimer levels (3.2 mg/dl, normal <0 href="https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3343255/figure/F2/" target="figure" class="fig-table-link figpopup" rid-figpopup="F2" rid-ob="ob-F2" co-legend-rid="lgnd_F2" xss=removed>Figure 2] and child was maintaining platelets above 90,000/μL. Neurological examination revealed features of hydrocephalous and global developmental delay. Steroids tapered gradually over 2 weeks and slow vincristine tapering was planned over the next 2-3 months. Ultrasound skull demonstrated post hemorrhagic hydrocephalous, for which ventriculoperitoneal shunt was advised. However, parents refused for surgery.
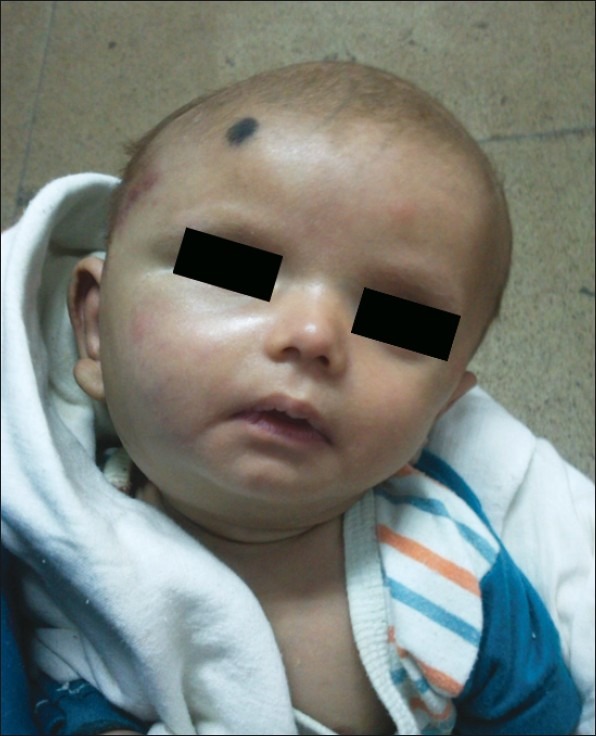
| Figure 2:After 3 month of therapy. Significant reduction in size of tumor
DISCUSSION
Kasabach and Merritt first reported the case of an infant with thrombocytopenic purpura due to what they believed to be a giant capillary hemangioma.[2] Thereafter, the association of a capillary hemangioma and thrombocytopenia was labeled Kasabach-Merritt syndrome (the name was later changed to KMP). The cardinal clinical and hematological features of this syndrome include an enlarging infantile vascular lesion, profound thrombocytopenia, a microangiopathic hemolytic anemia, and consumptive coagulopathy.[3] However, recent studies support an association of KMP with the vascular tumors (kaposiform hemangioendothelioma and tufted angioma).[1]
KMP typically has its onset in early infancy with a median age of onset of 5 weeks. In approximately 50% of cases, KMP was associated with a vascular tumor diagnosed at birth with 90% of published cases diagnosed before 1 year of age. Though hemangioma was commonly diagnosed in newborn period, presentation with KMP in early neonatal period is rare. In 3 case series, 2 (13.3%), 5 (13.5%) and 0 patients presented with KMP in neonatal period.[3–5] Common sites of lesion are chest and abdomen, and face has been less commonly reported site of hemangioma. Facial hemangioma was reported in 1 (6.6%), 5 (13.5%) and 0 cases in 3 studies.[3–5] Our child presented in early neonatal period with facial hemangioma, KMP and intracranial bleed.
All subjects with KMP had profound thrombocytopenia and hypofibrinogenemia with elevated fibrin split products (D-dimers), suggestive of an active consumptive coagulopathy. Platelet counts at the time of diagnosis ranged from 6000 to 98,000/μL with fibrinogen levels less than 100 mg/dL; whereas, D-dimers were always greater than 1. Evidence of intravascular hemolysis, including red blood cell fragmentation, elevated LDH, and hyperbilirubinemia, was a common finding.[1] Thrombocytopenia and consumption coagulopathy are usually caused by endothelial defects within the hemangioma causing platelet activation, platelet–fibrin thrombosis formation, consumption of clotting and coagulation factors and increased fibrinolysis.[6]
Magnetic resonance imaging (MRI) was the most frequently used modality to assess vascular tumors associated with KMP.[1]
There are two major treatment objectives; the control of the coagulopathy and thrombocytopenia as well as eradication of the hemangioma.[6] Classical treatment of the Kasabach-Merritt syndrome consists of tumor eradication. It can be achieved by different techniques: Surgery, radiation, embolization, and medical management. Surgery is recommended for single cutaneous lesions or multiple lesions in the spleen (splenectomy) or liver (wedge resection/hepatectomy). This is the only treatment that provides cure in significant number of cases. However, before each surgical intervention the patient must be stabilized.[7] The size of the hemangioma and its location often hinder surgical excision. Moreover, uncontrolled coagulopathy is a contraindication for surgery. Initially, radiotherapy was the most popular method to treat hemangiomas as they were considered to be very radiosensitive.[6] Radiation therapy often results in good hemangioma regression, but could be associated with local tissue damage and potential long-term risk of carcinogenesis.[8] Embolization can be performed with suitable vascular anatomy only.[7] When eradication is contraindicated or unfeasible, medical treatment is the only alternative. Consequently, different treatment regimens have been used with varying combination and success including systemic corticosteroids, irradiation, compression, embolization, antifibrinolytic agents, platelet aggregation inhibitors, interferon, chemotherapy as well as other strategies.[6] Some of the authors have suggested stepwise models for treatment of KMP in children;[4,5] however, there is no consensus on treatment. Steroids are used as first step in the treatment of KMS and other forms of invasive hemangiomas in most of the studies. The mechanisms of steroid therapy are unclear, but prednisone appears to increase vasoconstriction, inhibit fibrinolysis (by inhibiting production of tissue plasminogen activators and increasing the plasminogen activator inhibitors),[9] increase platelet longevity and disrupt angiogenesis. Possible complications of steroid use include interference with growth, reversible osteoporosis, and increased susceptibility to infection.[4] Different authors have reported varied response rate of steroids ranging from 11.4[4] to 66%.[10] Most patients responding to corticosteroids do so with a dose of prednisolone of 2-3 mg/kg/day within a few days. If a response is achieved, the dose is reduced slowly; too rapid a reduction in dose, particularly during the proliferative phase, is often associated with a recrudescence of symptoms. If no response is seen within a week or two after starting the therapy, then either the dose is increased or an alternative therapy is commenced.[10] Our child responded well to steroids initially; however, steroids therapy was disrupted by cholestasis, necessitating 2nd line therapy.
Interferon alpha has been used in some of the studies as second line therapy after failure of steroids. It inhibits angiogenesis and proliferation by suppressing over expression of basic fibroblast growth factor (bFGF), an angiogenic protein, in infantile hemagiomas. Adverse effects of interferon alpha-2b include constitutional symptoms (e.g., fever, malaise and fatigue, elevated liver enzymes, nausea, renal failure, transient neutropenia and anemia, hypothyroidism, bone marrow suppression, and myalgia) and neurological symptoms (e.g., gait abnormalities, memory disturbances, ataxia, paresthesia, numbness, oculomotor nerve paralysis, retinopathy, spastic diplegia).[6] Some of the serious adverse effects and high cost usually prohibit interferon use, particularly in developing world as in present child.
Vincristine is a naturally occurring vinca alkaloid. It interferes with the mitotic spindle microtubules by binding to tubulin, resulting in inhibition of mitosis. In vitro, vincristine also induces apoptosis of tumor cells as well as endothelial cells. Neurotoxicity is the dose-limiting side effect of vincristine. Treatment is administered at standard doses (1–1.5 mg/m2 or 0.05–0.065 mg/kg) in weekly intervals via central access and should be continued until a sustained increase in the platelet count has occurred. Average time reported for normalization of the platelet count was 5.3 weeks. Enjolras and associates have reported that several steroid non-responders show dramatic response to vincristine.[11] However, it is recommended that vincristine should be added when other modalities are unsuccessful.[7]
Vincristine has been frequently used in older children; however, its use in neonatal period has rarely been reported. Vincristine was used in only 2 neonates in a review on use of vincristine in KMP.[3] Our child was started on vincristine with steroids and responded well with normalization of coagulopathy and regression in size of vascular lesion.
Antifibrinolytic agents (e.g., E-aminocaproic acid or tranexamic acid) have been used in the management of KMP with variable responses. Antiplatelet agents (aspirin, dipyridamole) have also been used in some of the cases. It has been suggested that transfusion of exogenous blood products be withheld if there is no evidence of bleeding , as cytokines present in transfused blood may exacerbate the angiogenic process. Compression therapy has been reported to be useful in a number of patients with hemangiomas and is a simple adjunctive measure.[8]
In conclusion, neonatal KMP is a rare phenomenon. Early diagnosis and institution of treatment is associated with favorable outcome. Steroids are considered as the most effective 1st line treatment. Vincristine, interferon alpha, antifibrinolytics, radiation, embolization, and surgery are subsequent treatment options in steroid non-responders based on affordability, availability, and feasibility of a particular modality.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- Kelly M. Kasabach-Merritt Phenomenon. Pediatr Clin North Am 2010;57:1085-9.
- Kasabach HH, Merritt KK. Capillary hemangioma with extensive purpura: Report of a case. Am J Dis Child 1940;59:1063-70.
- Haisley-Royster C, Enjolras O, Frieden IJ, Garzon M, Lee M, Oranje A, et al. Kasabach-Merritt phenomenon: A retrospective study of treatment with vincristine. J Pediatr Hematol Oncol 2002;24:459-62.
- Shin HY, Ryu KH, Ahn HS. Stepwise multimodal approach in the treatment of Kasabach-Merritt syndrome. Pediatr Int 2000;42:620-4.
- Wananukul S, Nuchprayoon I, Seksarn P. Treatment of Kasabach-Merritt syndrome: A stepwise regimen of prednisolone, dipyridamole, and interferon. Int J Dermatol 2003;42:741-8.
- Hesselmann S, Micke O, Marquardt T, Baas S, Bramswig JH, Harms E, et al. Kasabach-Merritt syndrome: A review of the therapeutic options and a case report of successful treatment with radiotherapy and interferon alpha. Br J Radiol 2002;75:180-4.
- Abass K, Saad H, Kherala M, Elsayed AA. Successful treatment of Kasabach-Merritt syndrome with vincristine and surgery: A case report and review of literature. Cases J 2008;1:1.
- Blei F, Karp N, Rofsky N, Rosen R, Greco MA. Successful multimodal therapy for kaposiform hemangioendothelioma complicated by Kasabach-Merritt phenomenon: Case report and review of literature. Pediatr Hematol Oncol 1998;15:295-305.
- Dresse MF, David M, Hume H, Blanchard H, Russo P, Van Doesberg N, et al. Successful treatment of Kasabach Merritt syndrome with Prednisolone and epsilon aminocaproic acid. Pediatr Hematol Oncol 1991;8:329-34.
- ;Hall DG. Kasabach-Merritt syndrome: Pathogenesis and management. Br J Hematol 2001;112:851-62.
- Enjolras O, Mulliken JB, Wassef M, Frieden IJ, Rieu PN, Burrows PE, et al. Residual lesions after Kasabach-Merritt phenomenon in 41 patients. J Am Acad Dermatol 2000;42:225-35.
| Figure 1:At presentation. Hemangioma involving right half of face and earlobe
| Figure 2:After 3 month of therapy. Significant reduction in size of tumor
References
- Kelly M. Kasabach-Merritt Phenomenon. Pediatr Clin North Am 2010;57:1085-9.
- Kasabach HH, Merritt KK. Capillary hemangioma with extensive purpura: Report of a case. Am J Dis Child 1940;59:1063-70.
- Haisley-Royster C, Enjolras O, Frieden IJ, Garzon M, Lee M, Oranje A, et al. Kasabach-Merritt phenomenon: A retrospective study of treatment with vincristine. J Pediatr Hematol Oncol 2002;24:459-62.
- Shin HY, Ryu KH, Ahn HS. Stepwise multimodal approach in the treatment of Kasabach-Merritt syndrome. Pediatr Int 2000;42:620-4.
- Wananukul S, Nuchprayoon I, Seksarn P. Treatment of Kasabach-Merritt syndrome: A stepwise regimen of prednisolone, dipyridamole, and interferon. Int J Dermatol 2003;42:741-8.
- Hesselmann S, Micke O, Marquardt T, Baas S, Bramswig JH, Harms E, et al. Kasabach-Merritt syndrome: A review of the therapeutic options and a case report of successful treatment with radiotherapy and interferon alpha. Br J Radiol 2002;75:180-4.
- Abass K, Saad H, Kherala M, Elsayed AA. Successful treatment of Kasabach-Merritt syndrome with vincristine and surgery: A case report and review of literature. Cases J 2008;1:1.
- Blei F, Karp N, Rofsky N, Rosen R, Greco MA. Successful multimodal therapy for kaposiform hemangioendothelioma complicated by Kasabach-Merritt phenomenon: Case report and review of literature. Pediatr Hematol Oncol 1998;15:295-305.
- Dresse MF, David M, Hume H, Blanchard H, Russo P, Van Doesberg N, et al. Successful treatment of Kasabach Merritt syndrome with Prednisolone and epsilon aminocaproic acid. Pediatr Hematol Oncol 1991;8:329-34.
- ;Hall DG. Kasabach-Merritt syndrome: Pathogenesis and management. Br J Hematol 2001;112:851-62.
- Enjolras O, Mulliken JB, Wassef M, Frieden IJ, Rieu PN, Burrows PE, et al. Residual lesions after Kasabach-Merritt phenomenon in 41 patients. J Am Acad Dermatol 2000;42:225-35.