 PDF
PDF  Views
Views  Share
Share


A rare case of translocation (12;22) (p13;Q) in Ewings sarcoma
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2014; 35(01): 89-92
DOI: DOI: 10.4103/0971-5851.133730
Abstract
Cytogenetic or immunohistochemical studies are often required to differentiate Ewing′s sarcoma (ES) from other small round cell tumors. Herein we report a case of 13-year-old boy who presented with a large presacral lesion. Hemogram and biochemical parameters were normal except lactate dehydrogenase showing value of 96.40/IU/L, magnetic resonance imaging of the spine showed a large mass in presacral lesion (8 cm × 7 cm × 9 cm), with destruction of the sacrum (S2 S3 and S4) with interspinal extension. Bone scan showed multiple pelvic bone lesions, radiograph of chest, ultrasound of abdomen, pelvis and electrocardiogram were within normal limits. Bone marrow was not involved. Cells from the fine needle aspirate were cultured for short term using RPMI medium and karyotype obtained showed a t(12;22)(p12;q12) instead of the classic t(11;22). Diagnosis of ES was also confirmed by studies using immunohistochemistry for MIC2 which was positive, synaptophysin was inconclusive and leukocyte common antigen, desmin negative. This case provides evidence of the importance of chromosome 22, in the etiology of the disease.
Publication History
Article published online:
19 July 2021
© 2014. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Cytogenetic or immunohistochemical studies are often required to differentiate Ewing's sarcoma (ES) from other small round cell tumors. Herein we report a case of 13-year-old boy who presented with a large presacral lesion. Hemogram and biochemical parameters were normal except lactate dehydrogenase showing value of 96.40/IU/L, magnetic resonance imaging of the spine showed a large mass in presacral lesion (8 cm × 7 cm × 9 cm), with destruction of the sacrum (S2 S3 and S4) with interspinal extension. Bone scan showed multiple pelvic bone lesions, radiograph of chest, ultrasound of abdomen, pelvis and electrocardiogram were within normal limits. Bone marrow was not involved. Cells from the fine needle aspirate were cultured for short term using RPMI medium and karyotype obtained showed a t(12;22)(p12;q12) instead of the classic t(11;22). Diagnosis of ES was also confirmed by studies using immunohistochemistry for MIC2 which was positive, synaptophysin was inconclusive and leukocyte common antigen, desmin negative. This case provides evidence of the importance of chromosome 22, in the etiology of the disease.
The t(11;22)(q24;q12) is the most common translocation for ES and is present in more than 85% of cases.[1] This translocation leads to an EWS/FLI1 fusion gene in all cases. In a few instances, complex translocations, involving chromosomes 11 and 22 and a third chromosome or other variant translocations not involving chromosome 11 also have been reported.[2] They are molecularly characterized by expression of chimeric transcripts generated by specific chromosomal translocations, most commonly involving fusion of the EWS gene to a member of the Ewing tumors family of transcription factors, which include FLI1, ERG, ETV1, E1AF and FEV.[3] Sarcomas are a heterogeneous group of cancers derived from the connective tissue lineage. The etiology of these tumors is unknown, and the vast majority of the cases occur without known hereditary factors. In the last two decades, the finding of specific acquired chromosomal alterations in sarcomas has helped in many cases to understand the underlying genetic basis of this tumor.[4]
Ewing's sarcoma (ES), peripheral primitive neuroectodermal tumors (pPNETs) and Askin tumors are referred to as Ewing tumors (ETs). ES is the second most common malignant bone tumor in children and young adults, occurring with a male/female ratio of 1.5:1.[1,5] The most frequent primary site is the femur, followed by the pelvis arising from the ileum, ischium, pubic bone, or sacrum. Tumors may also originate in the tibia, fibula, or the bones of the feet. ES is the result of a translocation between chromosome 11 and 22, which fuses the EWS gene on chromosome 22 to the FLI1 gene on chromosome 11. EWS/FLI1 functions as the master regulator.[6]
A review of literature shows the chromosome partners involved in translocation with chromosome 22 in sarcomas are 1, 2, 4, 6, 7, 9, 11, 12, 17, 19, 20 and 21 as shown in the Chart 1. Nearly 85%-cases of ES contain the t(11;22)(q24;q12) Chromosomal translocation that encodes the EWS/FLI oncoprotein. Besides the t(11;22), however, many cases have otherwise simple karyotypes with no other demonstrable abnormalities. Furthermore, it seems that an underlying genetic susceptibility to ES, if it exists, must be rare.[7] Although Pro-B acute lymphoblastic leukemia showed t(12;22)(p13;q12) with EWSR1 → ZNF384 genes and expression of myeloid antigens, having relatively good prognosis,[8] the present case showed same translocation t(12;22)(p13;q12) but in ES.
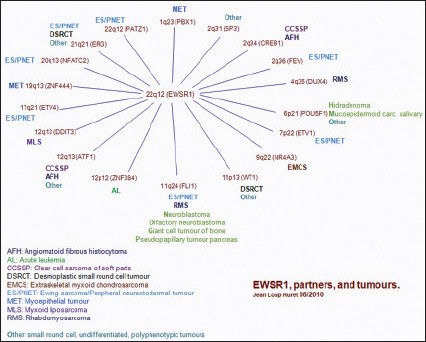
| Chart 1:EWSR1 partners and tumors
CASE REPORT
The case we present here is a 13-year-old boy who presented with the complaints of left lower limb weakness for 5 months and low back pain for 6 months. Hemogram and biochemical parameters were normal except lactate dehydrogenase (LDH) showing value of 96.40/IU/L, magnetic resonance imaging of the spine showed a large mass in presacral lesion (8 cm × 7 cm × 9 cm), with destruction of the sacrum (S2 S3 and S4) with interspinal extension. Bone scan showed multiple pelvic bone lesions, radiograph of chest, ultrasound of abdomen, pelvis and electrocardiogram were within normal limits. Bone marrow was not involved. Fine-needle aspiration cytology of the mass proved it to be a small round cell tumor. Immunohistochemistry for MIC2 was positive. Synaptophysin was inconclusive and leukocyte common antigen, desmin were negative [Figure [Figure1a1a and andbb].
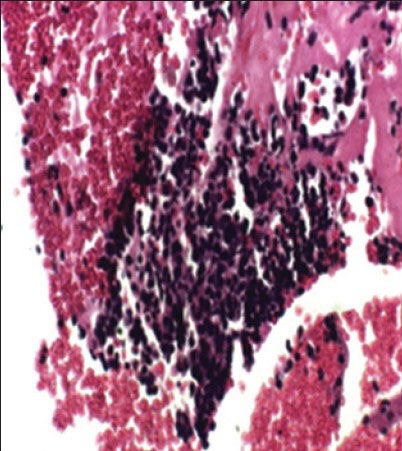
| Figure 1a:EWSR1 partners and tumorsCell block of Ewing's sarcoma (H and E)
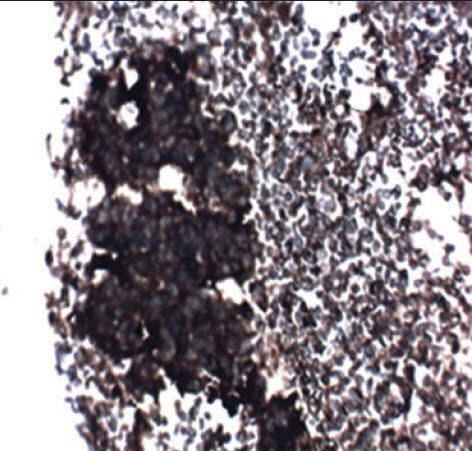
| Figure 1b:Cell block of Ewing's sarcoma positive for MIC2
CYTOGENETICS
Materials and methods
The fine-needle aspiration material obtained from the primary tumor mass was suspended in RPMI-1640 medium supplemented with 20% qualified fetal calf serum (GIBCO). Overnight cultures (16-18 h) were set up. The following day, Karyomax Colcemid solution (final concentration 0.05 μg/ml) was added for 30 min. Cells were subjected to hypotonic treatment (KCl-0.075 M) for 30 min at 37°C and fixed in methanol:acetic acid (3:1). A chromosome analysis was performed on GTG–banded metaphases and karyotype analysis [Figure [Figure2a2a and andb]b] was interpreted according to ISCN.[9]

| Figure 2a:GTG banded metaphase

| Figure 2b:GTG-banded karyotype showing 46, XY, t(12;22)(p13;q12). Arrows indicate break points
DISCUSSION
Typical ET can be diagnosed on morphology and immunohistochemistry, but the variants need cytogenetic or molecular confirmation of the diagnosis. ES and peripheral PNET strongly express the MIC2 (CD99) antigen in very high amounts which represents a highly selective and almost unique feature of these cells, therefore MIC2 analysis is a useful part of the panel of tests used in the differential diagnosis of ES/pPNET.[10] But the histological grade is of no prognostic significance, however, as all ES are of high grade. Fever, anemia and elevation of the number and values of white blood cell, erythrocyte sedimentation rate and LDH have been reported to indicate more extensive disease and a poorer prognosis.[1]
The incidence of classical t(11;22) in ES is around 90% approximately. Involvement of other chromosomes participating in translocation with 22 such as 21 is 5%, 2%, 4%, 7% and 17% are 1%, respectively and are rare [Table 1].
Table 1
The frequency of t(11;22) and its variants in ES

|
The present case adds on to this rare list of abnormalities in ES. The EWS-FLI1 translocation has been reported in two polyphenotypic tumors and two rhabdomyosarcomas.[11] Cytogenetic abnormalities are highly specific to ETs; chromosome 22 is most frequently involved in structural changes detected in cells of ES. Although t(11;22) is most frequent, chromosome 22 is also involved in other translocations in neoplasia, suggesting that the break point on chromosome 22 seen in ES cells may be a more important factor in the origin of this tumor than the loci to which the deleted segment is translocated.[1,5,6,12,13,14] In a case of ES which Whang-Peng et al.[15] examined it had a translocation involving chromosome 6 and 12, with a break point (12p13).[15] This finding with the absence of t(11;22) raises the possibilities that soft-tissue ES is a tumor significantly different in origin and pathogenesis from ES of the bone because our case presented with the tumor of the sacral bone. More cases need to be studied to determine the significance and incidence of this abnormality in ES. It is also important to determine whether additional structural chromosomal aberrations are present in ES tumors because it appears that a more complex karyotype with multiple chromosomal aberrations is associated with a poor outcome in ES.[16,17]
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- Iwamoto Y. Diagnosis and treatment of Ewing′s sarcoma. Jpn J Clin Oncol 2007;37:79-89.
- Desmaze C, Brizard F, Turc-Carel C, Melot T, Delattre O, Thomas G, et al. Multiple chromosomal mechanisms generate an EWS/FLI1 or an EWS/ERG fusion gene in Ewing tumors. Cancer Genet Cytogenet 1997;97:12-9.
- Ng TL, O′Sullivan MJ, Pallen CJ, Hayes M, Clarkson PW, Winstanley M, et al. Ewing sarcoma with novel translocation t(2;16) producing an in-frame fusion of FUS and FEV. J Mol Diagn 2007;9:459-63.
- Gabriela Mercado, Frederic Barr. Chromosomal Translocations in Sarcomas: New Perspectives ESUN (http://sarcomahelp.org/newsletter/february_2006.html). A periodical for sarcoma community, ESUN 2006;3:1
- Burt M, Karpeh M, Ukoha O, Bains MS, Martini N, McCormack PM, et al. Medical tumors of the chest wall. Solitary plasmacytoma and Ewing′s sarcoma. J Thorac Cardiovasc Surg 1993;105:89-96.
- Owen LA, Kowalewski AA, Lessnick SL. EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing′s sarcoma. PLoS One 2008;3:e1965.
- Toomey EC, Schiffman JD, Lessnick SL. Recent advances in the molecular pathogenesis of Ewing′s sarcoma. Oncogene 2010;29:4504-16.
- Gorello P, La Starza R, Mecucci C. t(12;22)(p13;q12). Atlas Genet Cytogenet Oncol Haematol. November 2007. (http://AtlasGeneticsOncology.org/Anomalies/t1222p13q12ID1371.html)
- ISCN. In: Mitelman F, editor. An International System for Human Cytogenetic Nomenclature. Basel: Karger; 1995.
- Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M. MIC2 is a specifi c marker for Ewing′s sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewing′s sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specifi c chromosome aberration. Cancer 1991;67:1886-93.
- Longtin R. Ewing′s sarcoma: A miracle drug waiting to happen? J Natl Cancer Inst 2003;95:1574-6.
- Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT. A second Ewing′s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet 1994;6:146-51.
- Jeon IS, Davis JN, Braun BS, Sublett JE, Roussel MF, Denny CT, et al. A variant Ewing′s sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene 1995;10:1229-34.
- Sumegi J, Nishio J, Nelson M, Frayer RW, Perry D, Bridge JA. A novel t(4;22)(q31;q12) produces an EWSR1-SMARCA5 fusion in extraskeletal Ewing sarcoma/primitive neuroectodermal tumor. Mod Pathol 2011;24:333-42.
- Whang-Peng J, Triche TJ, Knutsen T, Miser J, Kao-Shan S, Tsai S, et al. Cytogenetic characterization of selected small round cell tumors of childhood. Cancer Genet Cytogenet 1986;21:185-208.
- Zielenska M, Zhang ZM, Ng K, Marrano P, Bayani J, Ramirez OC, et al. Acquisition of secondary structural chromosomal changes in pediatric ewing sarcoma is a probable prognostic factor for tumor response and clinical outcome. Cancer 2001;91:2156-64.
- Wang L, Bhargava R, Zheng T, Wexler L, Collins MH, Roulston D, et al. Undifferentiated small round cell sarcomas with rare EWS gene fusions: Identifi cation of a novel EWSSP3 fusion and of additional cases with the EWS-ETV1 and EWS-FEV fusions. J Mol Diagn 2007;9:498-509.
| Chart 1:EWSR1 partners and tumors
| Figure 1a:EWSR1 partners and tumorsCell block of Ewing's sarcoma (H and E)
| Figure 1b:Cell block of Ewing's sarcoma positive for MIC2
| Figure 2a:GTG banded metaphase
| Figure 2b:GTG-banded karyotype showing 46, XY, t(12;22)(p13;q12). Arrows indicate break points
References
- Iwamoto Y. Diagnosis and treatment of Ewing′s sarcoma. Jpn J Clin Oncol 2007;37:79-89.
- Desmaze C, Brizard F, Turc-Carel C, Melot T, Delattre O, Thomas G, et al. Multiple chromosomal mechanisms generate an EWS/FLI1 or an EWS/ERG fusion gene in Ewing tumors. Cancer Genet Cytogenet 1997;97:12-9.
- Ng TL, O′Sullivan MJ, Pallen CJ, Hayes M, Clarkson PW, Winstanley M, et al. Ewing sarcoma with novel translocation t(2;16) producing an in-frame fusion of FUS and FEV. J Mol Diagn 2007;9:459-63.
- Gabriela Mercado, Frederic Barr. Chromosomal Translocations in Sarcomas: New Perspectives ESUN (http://sarcomahelp.org/newsletter/february_2006.html). A periodical for sarcoma community, ESUN 2006;3:1
- Burt M, Karpeh M, Ukoha O, Bains MS, Martini N, McCormack PM, et al. Medical tumors of the chest wall. Solitary plasmacytoma and Ewing′s sarcoma. J Thorac Cardiovasc Surg 1993;105:89-96.
- Owen LA, Kowalewski AA, Lessnick SL. EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing′s sarcoma. PLoS One 2008;3:e1965.
- Toomey EC, Schiffman JD, Lessnick SL. Recent advances in the molecular pathogenesis of Ewing′s sarcoma. Oncogene 2010;29:4504-16.
- Gorello P, La Starza R, Mecucci C. t(12;22)(p13;q12). Atlas Genet Cytogenet Oncol Haematol. November 2007. (http://AtlasGeneticsOncology.org/Anomalies/t1222p13q12ID1371.html)
- ISCN. In: Mitelman F, editor. An International System for Human Cytogenetic Nomenclature. Basel: Karger; 1995.
- Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M. MIC2 is a specifi c marker for Ewing′s sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewing′s sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specifi c chromosome aberration. Cancer 1991;67:1886-93.
- Longtin R. Ewing′s sarcoma: A miracle drug waiting to happen? J Natl Cancer Inst 2003;95:1574-6.
- Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT. A second Ewing′s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet 1994;6:146-51.
- Jeon IS, Davis JN, Braun BS, Sublett JE, Roussel MF, Denny CT, et al. A variant Ewing′s sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene 1995;10:1229-34.
- Sumegi J, Nishio J, Nelson M, Frayer RW, Perry D, Bridge JA. A novel t(4;22)(q31;q12) produces an EWSR1-SMARCA5 fusion in extraskeletal Ewing sarcoma/primitive neuroectodermal tumor. Mod Pathol 2011;24:333-42.
- Whang-Peng J, Triche TJ, Knutsen T, Miser J, Kao-Shan S, Tsai S, et al. Cytogenetic characterization of selected small round cell tumors of childhood. Cancer Genet Cytogenet 1986;21:185-208.
- Zielenska M, Zhang ZM, Ng K, Marrano P, Bayani J, Ramirez OC, et al. Acquisition of secondary structural chromosomal changes in pediatric ewing sarcoma is a probable prognostic factor for tumor response and clinical outcome. Cancer 2001;91:2156-64.
- Wang L, Bhargava R, Zheng T, Wexler L, Collins MH, Roulston D, et al. Undifferentiated small round cell sarcomas with rare EWS gene fusions: Identifi cation of a novel EWSSP3 fusion and of additional cases with the EWS-ETV1 and EWS-FEV fusions. J Mol Diagn 2007;9:498-509.