 PDF
PDF  Views
Views  Share
Share


Adrenocortical Carcinoma: A Rare Tumor in a 12-year-old Girl
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2018; 39(04): 533-535
DOI: DOI: 10.4103/ijmpo.ijmpo_128_17
Abstract
A 12-year-old girl presented with intra-abdominal mass and cushingoid features. On investigation, she was diagnosed as a case of functioning adrenocortical carcinoma. Two cycles of neoadjuvant chemotherapy followed by excision of mass with right nephrectomy was performed. On 6-month follow-up, recurrence and metastasis were identified which were managed with surgery and chemotherapy.
Publication History
Article published online:
17 June 2021
© 2018. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
A 12-year-old girl presented with intra-abdominal mass and cushingoid features. On investigation, she was diagnosed as a case of functioning adrenocortical carcinoma. Two cycles of neoadjuvant chemotherapy followed by excision of mass with right nephrectomy was performed. On 6-month follow-up, recurrence and metastasis were identified which were managed with surgery and chemotherapy.
Keywords
Adrenocortical carcinoma - cushingoid - pediatricIntroduction
Adrenocortical carcinoma (ACC) is one of the rare tumors with worldwide incidence of 0.3–0.38/million/year in children.[1] [2] The first case was reported in 1865.[3] Nearly 95% of pediatric ACCs are functional as opposed to adults where they are mostly nonfunctional.[4] Virilization is the most common presentation followed by Cushing’s syndrome.[5] Surgical resection is the mainstay of treatment. Even for patients with resected tumors, the median survival is only 32 months.[5]
Case Report
A 12-year-old girl presented with the complaints of abdominal pain and swelling for 1 month. The patient was hypertensive and started on antihypertensives. She had cushingoid features such as moon face and mild clitoromegaly and a large lump in the right hypochondrium extending to the adjacent quadrants, firm in consistency with ill-defined upper margin. On investigations, basal cortisol was 16.15 mcg/dl (normal range - 7–8 mcg/dl at 8:30 am) and dihydroepiandrosterone (DHEAS) was 970 mcg/dl (normal range - 8.6–169.8 mcg/dl). Urinary vanillylmandelic acid and metanephrines were normal. Ultrasound (USG) abdomen showed a highly vascular, well-defined hypoechoic lesion in segments VI, VII, and VIII of liver measuring 16 cm × 14 cm. Contrast-enhanced computed tomography abdomen (CECT) revealed a heterogeneously enhancing highly vascular mass of 14.5 cm × 12.4 cm × 15.3 cm size in the right adrenal fossa with specs of calcification, displacing kidney inferiorly and medially [Figure 1]. The right adrenal gland was indistinguishable from the mass. Inferior vena cava (IVC) was compressed anteromedially, and fat planes between the mass and liver and the right kidney were obliterated. Meta-iodobenzylguanidine (MIBG) scan showed no focus of tracer uptake [Figure 2]. Flourodeoxyglucose-positron emission tomography (FDG-PET) showed a metabolically active mass with calcification in the right suprarenal region [Figure 3]. With a diagnosis of highly vascular ACC, two cycles of neoadjuvant chemotherapy with cisplatin, etoposide, and cyclophosphamide were given with the aim of reducing vascularity and size. Radiologically, there was no change in size, so the decision of excision was made. On exploration, a huge mass about 15 cm × 14 cm × 18 cm with large vessels traversing over its surface was found in the right adrenal region. Plane between the liver and mass was very vascular, but the mass could be dissected off the liver. IVC was stretched and draped over the tumor, but could be dissected off. There was no plane between the mass and the right kidney [Figure 4]. Excision of the right adrenal mass with right nephrectomy was done [Figure 5]. There was no intraoperative spillage. Postoperatively, the patient was gradually weaned off antihypertensives. Steroids were given for 15 days. Histopathological report confirmed the diagnosis of ACC. Margins were clear. Modified Weiss criteria score was 5/9.[6] Follow-up, serum DHEAS, and cortisol were normal. At 6-month follow-up, the child was asymptomatic, but repeat serum DHEAS was elevated. The FDG-PET suggested a solitary lesion in the right lobe of the liver and peritoneal nodules. The patient underwent radiofrequency ablation (RFA) of liver lesion. However, follow-up FDG-PET after RFA showed increase in the size of peritoneal metastasis. The patient underwent peritoneal metastasectomy and was started on tablet mitotane. On follow-up after 6 months, she is asymptomatic and normotensive.
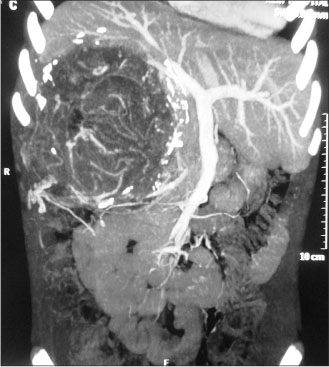
| Figure 1:Contrast-enhanced computed tomography image of tumor
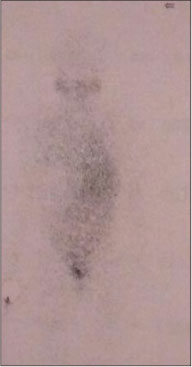
| Figure.2:Meta-iodobenzylguanidine scan
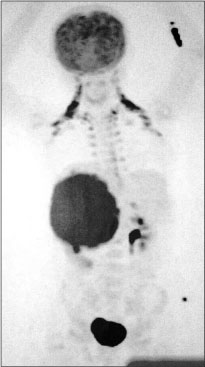
| Figure.3:Flourodeoxyglucose-positron emission tomography image
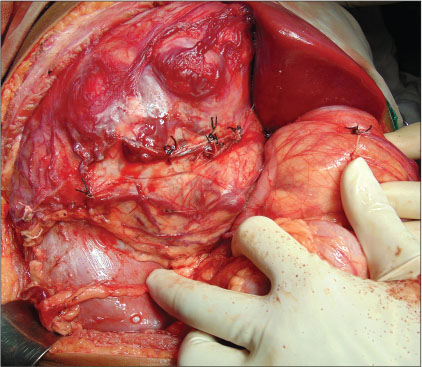
| Figure.4:Intraoperative image

| Figure.5:Gross specimen
Discussion
ACC is a very rare tumor in children, most of which are functional. Cushing’s syndrome is more common when tumor size is >10 cm. The index case had both features.
Laboratory evaluation of suspected case includes measurement of urinary 17-ketosteroid, 17-hydroxyprogesterone, free cortisol, DHEAS, testosterone, aldosterone, renin activity, and other 17-deoxysteroid precursors. They are useful indicators for recurrence also. USG is an important imaging modality for evaluating tumor site, size, extent, echogenicity, and extension into the IVC and right atrium. CECT remains the modality of choice for initial evaluation showing the presence of calcification, peripheral enhancement, central necrosis/hemorrhage, and distant metastases. To differentiate between pheochromocytoma and ACC, MIBG and FDG-PET scans are done. MIBG tracer uptake is present in the former; it was absent in the index case. FDG-PET scan differentiates between benign and malignant lesions.
The management of patients with ACC requires a multidisciplinary approach.
Surgical excision is the mainstay of treatment for resectable tumors. Medical management aims to control tumor growth and symptoms of hormonal excess. Mitotane has remained the only available adrenolytic agent, although response rates are modest at approximately 30%-and it is very costly. Patients with unresectable or recurrent tumors require palliative therapy in the form of cisplatin-based chemotherapy/radiotherapy, though results are disappointing.[7] [8] Systemic therapies for advanced ACC are limited, and there remains a scope for new and effective treatment.
According to the WHO classification, ACC is staged as Stages I and II as localized tumors ≤5 cm and >5 cm, respectively. Locally invasive tumors or regional lymph node metastases are Stage III and those invading adjacent organs or with distant metastases are Stage IV. In children, Stage I and II tumors are those completely excised with negative margins and with a tumor weight >200 g being Stage II. Stage III has microscopic or macroscopic residual disease or inoperable tumors, while Stage IV has distant metastasis. Poor prognostic factors are presentation after 5 years of age, tumor size >10.5 cm, tumors >400 g, necrosis, and frequent and atypical mitotic bodies. Stage at presentation is a significant prognostic factor.[2] [3] [4] [5] The overall 5-year survival rate is 54.2%. For Stages I to IV tumors, the approximate 5-year survival rate was 30%–45%, 12.5%–57%, 5%–18%, and 0%, respectively.[6] Surgical resection is the only therapy that significantly prolongs survival. Median survival in patients with unresectable tumors is 3–9 months, whereas after complete resection, 13–28 months. Modified Weiss criteria is useful for prognosis, which includes nine criteria. A score of 3 or more suggests malignancy.[8] About 50%–80%-patients relapse or progress to metastasis even after R0 resection. The index patient was Stage II and her modified Weiss criteria score was 5, but had poor prognostic factors – age >5 years, tumor size >10.5 cm, weighing >400 g, and had necrosis. Though complete excision of tumor with clear margins was possible, there was recurrence and metastasis.
Conclusion
ACC is a very rare pediatric tumor. Surgery is the mainstay of treatment and multidisciplinary approach is recommended in pediatric population.
Conflict of Interest
There are no conflicts of interest.
References
- Stiller CA. International variations in the incidence of childhood carcinomas. Cancer Epidemiol Biomarkers Prev 1994; 3: 305-10
- Allolio B, Fassnacht M. Clinical review: Adrenocortical carcinoma: Clinical update. J Clin Endocrinol Metab 2006; 91: 2027-37
- Anonymous. General melasma and short hair over the entire body of a child of three years, with conversion of the left supra-renal capsule into a large malignant tumor; The external organs of generation resembling that of adult life. Lancet 1865; 1: 175
- Michalkiewicz E, Sandrini R, Figueiredo B, Miranda EC, Caran E, Oliveira-Filho AG. et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the international pediatric adrenocortical tumor registry. J Clin Oncol 2004; 22: 838-45
- Ng L, Libertino JM. Adrenocortical carcinoma: Diagnosis, evaluation and treatment. J Urol 2003; 169: 5-11
- Mendonca BB, Lucon AM, Menezes CA, Saldanha LB, Latronico AC, Zerbini C. et al. Clinical, hormonal and pathological findings in a comparative study of adrenocortical neoplasms in childhood and adulthood. J Urol 1995; 154: 2004-9
- Veytsman I, Nieman L, Fojo T. Management of endocrine manifestations and the use of mitotane as a chemotherapeutic agent for adrenocortical carcinoma. J Clin Oncol 2009; 27: 4619-29
- Berruti A, Fassnacht M, Baudin E, Hammer G, Haak H, Leboulleux S. et al. Adjuvant therapy in patients with adrenocortical carcinoma: A position of an international panel. J Clin Oncol 2010; 28: e401-2
Address for correspondence
Publication History
Article published online:
17 June 2021
© 2018. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2,
Noida-201301 UP, India
| Figure 1:Contrast-enhanced computed tomography image of tumor
| Figure.2:Meta-iodobenzylguanidine scan
| Figure.3:Flourodeoxyglucose-positron emission tomography image
| Figure.4:Intraoperative image
| Figure.5:Gross specimen
References
- Stiller CA. International variations in the incidence of childhood carcinomas. Cancer Epidemiol Biomarkers Prev 1994; 3: 305-10
- Allolio B, Fassnacht M. Clinical review: Adrenocortical carcinoma: Clinical update. J Clin Endocrinol Metab 2006; 91: 2027-37
- Anonymous. General melasma and short hair over the entire body of a child of three years, with conversion of the left supra-renal capsule into a large malignant tumor; The external organs of generation resembling that of adult life. Lancet 1865; 1: 175
- Michalkiewicz E, Sandrini R, Figueiredo B, Miranda EC, Caran E, Oliveira-Filho AG. et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the international pediatric adrenocortical tumor registry. J Clin Oncol 2004; 22: 838-45
- Ng L, Libertino JM. Adrenocortical carcinoma: Diagnosis, evaluation and treatment. J Urol 2003; 169: 5-11
- Mendonca BB, Lucon AM, Menezes CA, Saldanha LB, Latronico AC, Zerbini C. et al. Clinical, hormonal and pathological findings in a comparative study of adrenocortical neoplasms in childhood and adulthood. J Urol 1995; 154: 2004-9
- Veytsman I, Nieman L, Fojo T. Management of endocrine manifestations and the use of mitotane as a chemotherapeutic agent for adrenocortical carcinoma. J Clin Oncol 2009; 27: 4619-29
- Berruti A, Fassnacht M, Baudin E, Hammer G, Haak H, Leboulleux S. et al. Adjuvant therapy in patients with adrenocortical carcinoma: A position of an international panel. J Clin Oncol 2010; 28: e401-2