 PDF
PDF  Views
Views  Share
Share


Ataxia telangiectasia: A report of two cousins and review of literature
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2011; 32(04): 217-222
DOI: DOI: 10.4103/0971-5851.95145
Abstract
Ataxia telangiectasia (AT) is a rare multisystem, neurodegenerative genetic disorder. Due to its wide clinical heterogeneity, it often leads physicians to an incorrect or missed diagnosis, and insight into this rare disease is important. Here is a case report of two cousins from the same family who showed salient characteristic features of AT along with the incidental finding of co-inheritance of hemoglobin E trait. Though both of them were from the same family, they showed differences in the type of humoral immune deficiencies, laboratory findings, and their susceptibility to develop different types of malignancies. One of them developed T cell acute lymphoblastic leukemia, isolated immunoglobulin A deficiency, and normal serum carcinoembryonic antigen (CEA) and carbohydrate antigen 19.9 (CA 19.9) levels. He expired at the age of nine years. The other, though a year older, has still got normal blood counts, normal immunoglobulin levels, and elevated serum CEA and CA 19.9 levels. Thus, insight into this disease is very important as AT patients require protection from unnecessary exposure to ionizing radiation to prevent malignancies. Diagnosis of AT allows appropriate genetic counseling for the family.
Keywords
Ataxia telangiectasia - hemoglobin E - T cell acute lymphoblastic leukemia - humoral immunodeficiencyPublication History
Article published online:
06 August 2021
© 2011. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Ataxia telangiectasia (AT) is a rare multisystem, neurodegenerative genetic disorder. Due to its wide clinical heterogeneity, it often leads physicians to an incorrect or missed diagnosis, and insight into this rare disease is important. Here is a case report of two cousins from the same family who showed salient characteristic features of AT along with the incidental finding of co-inheritance of hemoglobin E trait. Though both of them were from the same family, they showed differences in the type of humoral immune deficiencies, laboratory findings, and their susceptibility to develop different types of malignancies. One of them developed T cell acute lymphoblastic leukemia, isolated immunoglobulin A deficiency, and normal serum carcinoembryonic antigen (CEA) and carbohydrate antigen 19.9 (CA 19.9) levels. He expired at the age of nine years. The other, though a year older, has still got normal blood counts, normal immunoglobulin levels, and elevated serum CEA and CA 19.9 levels. Thus, insight into this disease is very important as AT patients require protection from unnecessary exposure to ionizing radiation to prevent malignancies. Diagnosis of AT allows appropriate genetic counseling for the family.
INTRODUCTION
Ataxia telangiectasia (AT) is a rare multisystem, neurodegenerative genetic disorder.[1] Due to its wide clinical heterogeneity, it often leads physicians to an incorrect or missed diagnosis, and insight into this rare disease is important. Here is a case report of two cousins from the same family who showed salient characteristic features of AT along with the incidental finding of co-inheritance of hemoglobin E (HbE) trait. Though both of them were from the same family, they showed differences in the type of humoral immune deficiencies, laboratory findings, and their susceptibility to develop different types of malignancies.
CASE REPORTS
Case 1
A 9-year-old male Muslim child came to the pediatric outpatient department (OPD) with the chief complaints of fever, multiple swellings in the neck and groin region, and loss of appetite for 10 days. Fever was intermittent, low grade and associated with sweating.
His past history of 9 years was significant. He was an offspring of a consanguineous marriage, a full term normal vaginally delivered baby with normal developmental milestones till the age of 9 months. There is history of recurrent diarrhea since the age of 9 months, meningitis at the age of 1.5 years, redness of eyes since the age of 2 years, recurrent ear discharge since the age of 4 years, and recurrent chest infections since the age of 6 years. Patient was symptomatically treated for all these recurrent infections. Meanwhile, all the investigations were performed. His complete hemogram showed variable degree of anemia, with normal white cell counts, platelets and a normal peripheral smear. High performance liquid chromatography for hemoglobin variant analysis showed heterozygous inheritance of hemoglobin E (HbA2 + E levels-26.4%, HbA0-62.4% and HbF-0.7%). The family studies showed that his father also had inheritance of HbE (heterozygous) as above, but his mother was normal.
His serum immunoglobulin A (IgA) was markedly reduced to 2 mg/dl (normal-35-250 mg/dl), however, tissue transglutaminase IgA antibody was normal, ruling out celiac disease. Serum alfa feto protein (AFP) was elevated to 67.78 IU/ml (normal-<5>
He had progressive ataxia since the age of 1 year and redness of eyes since 2 years of age. With all the above clinical and laboratory findings, he was diagnosed as AT with heterozygous inheritance of hemoglobin E disease at the age of 2 years.
On present examination, his vitals were stable. He had ocular telangiectasia, but no cutaneous involvement. He had cervical lymphadenopathy involving both right and left vertical chain, measuring approximately 8 × 10 cm, mobile, and non tender. Bilateral inguinal lymph nodes were palpable measuring 2 × 2 cm, free, mobile, and non tender. His height and weight was less than 50th percentile of normal. His developmental milestones were delayed. Chest examination revealed bilateral crepitations. Cardiovascular examination was normal. On abdominal examination liver was soft, palpable 2 cm below right mid-clavicular line. Spleen was not palpable. Central nervous system (CNS) examination showed a conscious, oriented child, but cerebellar signs (ataxia with inability to walk, intention tremor, nystagmus, dysdiadochokinesia, slurred speech) were present. Other CNS examination was unremarkable. Fundus was normal.
Routine hemogram showed mild anemia with leucocytosis (total leucocyte count-48,500 cells/cumm). Peripheral smear showed blasts for the first time, comprising 55% of the total leucocytes. Platelets were adequate. These blasts were 2-3 times larger than small lymphocytes with high nuclear-cytoplasmic ratio, opened up chromatin, nuclear cleaving, and scanty cytoplasm [Figure 1a].
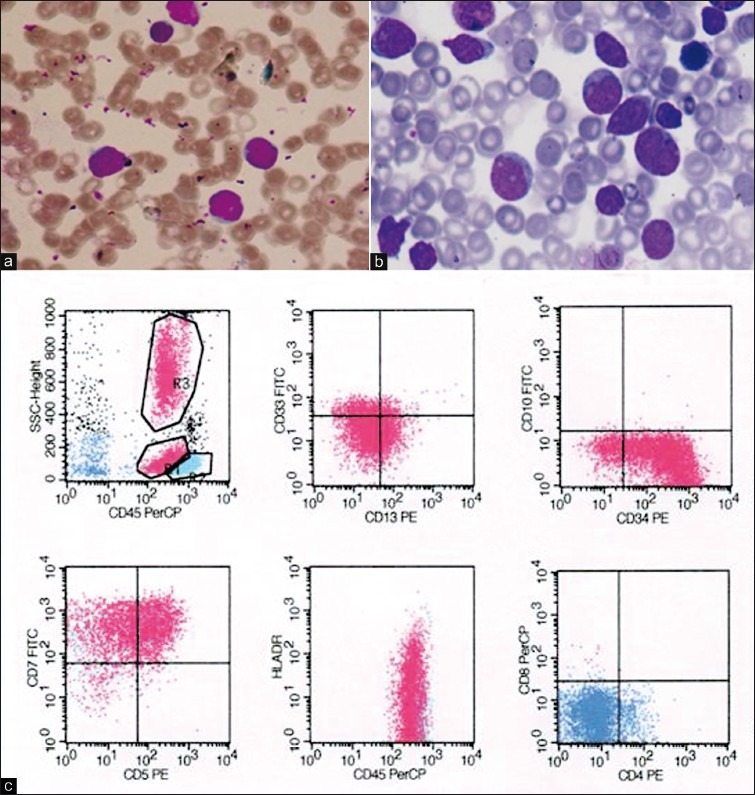
| Figure 1:Case 1 (a) Peripheral smear showing leucocytosis with blast population; (b) Bone marrow showing predominantly of blast population; (c) Immunophenotyping showing positivity for T-lymphoid lineage markers (CD7, CD5) and early stem cell marker (CD34, human leucocyte antigen [HLA]-DR). CD4 and CD8 are negative
Bone marrow aspiration was done. Smears were cellular showing predominantly a blast cell population with the morphology as described in peripheral smear. Few cells of myeloid and erythroid series were seen in different stages of maturation. Megakaryocytes were adequate and functioning and a possibility of acute leukemia was considered [Figure 1b].
Immunophenotyping was done on four color flow cytometer which showed strong positivity for T-lymphoid lineage markers (cluster of differentiation [CD] CD7, CD2, CD5, CD4, CD8) with negativity for B lineage markers [Figure 1c].
Fine needle aspiration cytology from all lymph nodes showed a picture suggestive of acute lymphoblastic leukemia/lymphoma.
Thus, a final diagnosis of T-acute lymphoblastic leukemia was made.
Chromosomal breakage studies were performed showing increased chromosomal rearrangements [Figure 2].
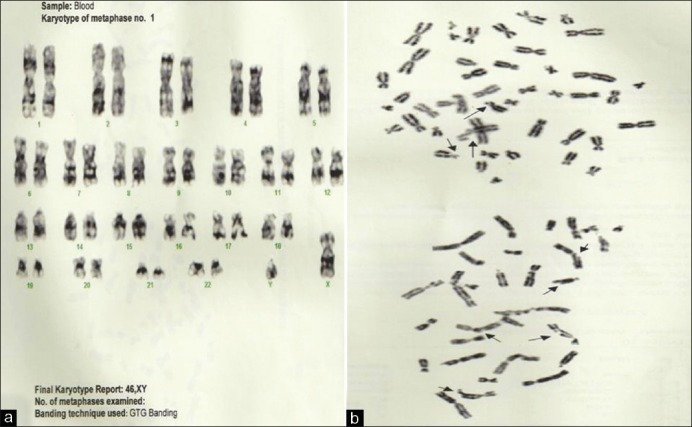
| Figure 2:Case 1 (a) Final karyotype report: 46, XY; (b) Chromosome rearrangement seen in two partial metaphases
Chemotherapy was started with vincristine (1 mg in 100 ml dextrose 5%), intravenous hydrocortisone (16 mg) intrathecal, cytosine arabinoside (30 mg) intrathecal, adriamycin (35 mg) intravenous once weekly and L-asparginase (7000 IU) intramuscular twice weekly. Patient was monitored with blood counts twice a week. Repeat bone marrow aspiration done after 4 weeks of initiation of chemotherapy showed 9% residual blast cells on immunophenotyping. Chemotherapy was continued. However, the child left the hospital against medical advice. On follow-up, it was found that the child expired a few days later after leaving the hospital.
Case 2
This child, aged 10 years is a cousin of case 1 [Figure 3]. He was a full term normal vaginally delivered child with recurrent multiple episodes of chest infections and gastro intestinal infection and cyanotic spells. He had global developmental retardation with seizure disorder and delayed speech. A past history of tuberculosis at 6 years of age was elicited, for which antitubercular therapy was given. Like his cousin, he had gradual development of ataxia with reddening of eyes.
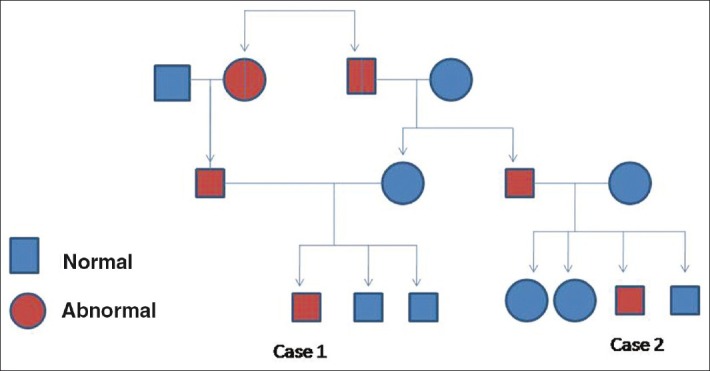
| Figure 3:Family tree showing the relationship between the two cases
On examination, patient had microcephaly with ataxia and speech defect. His vitals were stable. Chest, abdominal and cardiovascular examinations were normal. There was no organomegaly and lymphadenopathy. His developmental milestones were delayed and his height and weight was less than 50th percentile of normal.
His hemogram showed mild anemia, normal leucocytes, and adequate platelets. No abnormal cells were seen. Serum haptoglobin, mean corpuscular volume (MCV), and osmotic fragility were all reduced. Hemoglobin variant analysis showed heterozygous inheritance of hemoglobin E as seen in the index case. Serum ferritin, vitamin B12, and folic acid were within normal limits. Levels of IgA, IgG and IgM were within normal limits. Serum alpha-fetoprotein (AFP) was markedly elevated to 677.9 (<5 href="https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3343250/figure/F4/" target="figure" class="fig-table-link figpopup" rid-figpopup="F4" rid-ob="ob-F4" co-legend-rid="lgnd_F4" xss=removed>Figure 4].
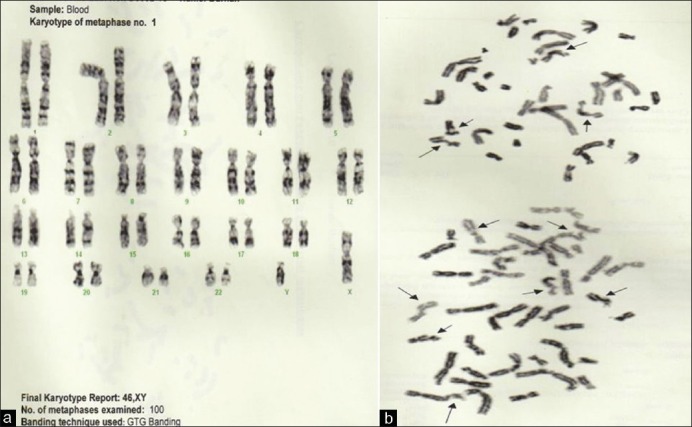
| Figure 4:Case 2 (a) Final karyotype report 46; (b): Chromosome rearrangement seen in two partial metaphases
Similar to his cousin, he is also a case of AT with heterozygous inheritance of hemoglobin E. This child is under follow-up.
DISCUSSION
The initial clinical description of ataxia telangiectasia (A-T) was reported by Syllaba and Henner in 1926, and the syndrome was first signed to the term “AT” by Boder and Sedgwick in 1957.[1] The prevalence of AT disease has estimated to be one case in 40,000 to one case in 100,1000.[1]
AT is a rare, autosomal recessive, multisystem disorder characterized by progressive cerebellar ataxia, pathognomic oculo-cutaneous telangiectasia, recurrent sinopulmonary infection, variable humoral and cellular immunodeficiency, a high incidence of malignancy, and hypersensitivity to ionizing radiation. High serum AFP, retardation of somatic growth, gonadal dysgenesis, and defective cell cycle check points are also seen.[1]
Though unrelated to each other, the incidental association of autosomal recessive HBE trait in a case of AT is a unique finding. According to our knowledge, no such association has been reported in the literature till now. This may be due to simultaneous inheritance of both autosomal recessive disorders together. Though, studies on association of AT with Fanconi anemia and autoimmune anemia are documented.[2,3]
Neurological symptoms are one of the earliest features of AT, most often in the form of cerebellar ataxia which is the clinical hallmark of AT and is present in all the patients.[1] Folgori et al., reported the case of a 3-year-old child affected by AT who presented exclusively with cutaneous granulomatosis and severe combined immunodeficiency, without neurological abnormalities.[4] Loeb et al., reported two children with a lymphoid malignancy who were later diagnosed as AT and the neurological manifestations of AT were misinterpreted as toxicity of the chemotherapy.[5]
Gatti RA et al., reported that ataxia typically becomes evident shortly after the child begins to walk (at about 12-14 months) of age and the movement disorders progress to the stage of enforcing a wheel chair existence by the age of 10-11 years as independent walking becomes impossible.[1] The ataxia is predominantly truncal ataxia at 1 year of age.[1] Both patients in our study developed progressive cerebellar ataxia and they were unable to walk by the age of 2 years.
The essential and pathognomic manifestation of this disease is ocular telangiectasia and is usually observed in patients between 3 and 6 years of age.[1] This pathognomic feature was also seen in both of our cases at the age of 2 years.
Impaired immunological status is a finding in almost all AT patients and decreased concentration or absence of serum IgA and IgE secondary to reduced synthesis of hypercatabolism has been reported in most patients.[1] Selective IgA deficiency is found in 50-80% of affected individuals. A variety of other humoral immunodeficiencies have been described, including IgG4 subclass levels. Antibody response to viral and bacterial antigens have been reported to be impaired.[1]
Similarly, in case no. 1, isolated IgA deficiency was found. However, in case no. 2, even though he had recurrent attacks of chest and gastrointestinal infections, his immunoglobulin levels were normal. This finding suggests that genetic heterogeneity is broad in AT representing a complex spectrum of the disease shaped by the genetic mutations, host characteristics, and environmental factors.[4]
In an Indian series of 22 patients of AT, IgG was normal in all patients, but IgM was low in 2 of 18 patients tested. Secretory IgA was absent in all patients.[6] Due to variable degree of immunodeficiency in AT patients, some patients present with recurrent severe infections, whereas others suffer from only mild infections.[1] Staples et al., reported that AT patients with no ataxia telangiectasia mutated (ATM) kinase activity had markedly more severe immunological phenotype than those expressing low levels of ATM activity.[7]
As AT has a wide clinical heterogeneity, there should be a high index of suspicion for AT among children with recurrent infections and immunodeficiency.[4] Accordingly, screening with serum AFP, which is a simple, rapid and reliable test for AT, should be a part of the evaluation of children with any congenital immunodeficiency. Both the above cases had elevated serum AFP levels. Elevated CEA was found in case 2 only.
More than 1/3rd of AT patients develop malignancy.[1] The most common type of malignancy is lymphoma, usually, the B-cell type.[8] Loss of heterogenity at the ATM gene and ATM protein deficiency have been described in acute lymphoblastic leukemia (ALL), B-CLL, T-cell prolymphocytic leukemia (T-PLL) and non-Hodgkin lymphoma (NHL), possibly representing major determinants for the prognosis of these disorders.[9] There is a 4-5 fold increased frequency of T cells tumor compared to B cell tumors in these patients.[9]
Tolenado and Lange et al., considered that AT patients (children) with ALL generally showed an unusually high proportion of unfavorable prognostic characteristics, like presentation at an older age (median 9 years), male predominance, higher initial white blood cell (WBC) count and occasionally presence of a mediastinal mass.[10]
In case no. 1, patient had normal WBC and morphology, till the age of 8 years, when blasts appeared in the peripheral blood. Case no. 2, who is a year older to case no. 1 has still got normal WBC counts, and is under strict follow-up.
Taylor et al., documented that there are several indicators of genetic heterogenity in AT suggesting that not all AT patients are equally susceptible to all T cell tumor types. Failure to diagnose AT in an infant with lymphoma/leukemia, may result in radiation therapy with conventional dosages, which is contraindicated in ATM patients.[8] The diagnosis of AT should be considered in all children with neuromotor dysfunction; particularly those who develop lymphoid malignancies. The consequences of missing the diagnosis may be detrimental, as radiation therapy and radiomimetic drugs should be avoided in patients with AT.[11]
It is also documented that non lymphoid tumors, mainly carcinomas represent about 13-22% of all malignancies in AT patients.[10] (Taylor et al.,) Concordance for tumor types within individual families suggest that particular gene defects may be associated with particular tumor type.[10] He suggested that AT gene in homozygous state may play a general role in the predisposition to cancer in many cells. Non lymphoid malignancies are usually seen in post adolescent AT patients[6] and may include solid tumors like ovarian, uterine, stomach, liver, and parotid gland tumors.[10]
Though there is no evidence of any non lymphoid tumor in case no. 2, but his elevated serum CEA and CA19.9 levels are alarming, as these tumor markers are found elevated in various epithelial malignancies. Thus, a strict follow-up is mandatory as he is already predisposed to develop an epithelial malignancy later in life.
The most common misdiagnosis in AT patients is cerebral palsy. AT in later stages can also be confused with Friedreich ataxia.[1] Elevated AFP levels clearly distinguish AT from other ataxias and immunodeficiency syndromes.[12]
CONCLUSION
Both these cousins showed salient characteristic features of AT along with the coincidental finding of co-inheritance of HbE trait. Though both are from the same family, genetic and clinical heterogeneity in AT is obviously evident from these cases. One of them developed T-ALL, isolated IgA deficiency and normal serum CEA and CA 19.9 levels. He expired at the age of 9 years. The other, though a year older, has still got normal blood counts, normal immunoglobulin levels, and elevated serum CEA and CA 19.9 levels. Thus, insight into this disease is very important as AT patients require protection from unnecessary exposure to ionizing radiation to prevent malignancies. Diagnosis of AT allows appropriate genetic counseling for the family.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- Huang KY, Shyur SD, Wang CY, Shen EY, Liang DC. Ataxia telangiectasia: Report of two cases. J Microbiol Immunol Infect 2001;34:71-5.
- Muller H. Recessively inherited deficiencies predisposing to cancer. Anticancer Res 1990;10:513-8.
- Kutukcluer N, Aksu G. Is there an association between autoimmune hemolytic anemia and ataxia-telangiectasia? Autoimmunity 2000;32:145-7.
- Folgori L, Scarselli A, Angelino G, Ferrari F, Antoccia A, Chessa L, et al. Cutaneous granulomatosis and combined immunodeficiency revealing Ataxia-Telangiectasia: A case report. Ital J Pediatr 2010;36:29.
- Loeb DM, Lederman HM, Winkelstein JA. Lymphoid malignancy as a presenting sign of ataxia-telangiectasia. J Paediatr Hematol Oncol 2000;22:464-7.
- Goyal V, Behari M. Dystonia as presenting manifestation of Ataxia Telangiectasia: A case report. Neurol India 2002;50:187-9.
- Staples ER, Mc Dermott EM, Reiman A, Byrd PJ, Ritchie S, Taylor AM, et al. Immunodeficiency in ataxia telangiectasia is correlated strongly with the presence of two null mutations in the ataxia telangiectasia mutated gene. Clin Exp Immunol 2008;153:214-20.
- Gatti RA. Ataxia Telangiectasia. Dermatol Clin 1995;13:1-6.
- Viniou N, Terpos E, Rombos J, Vaiopoulos G, Nodaras K, Stamatopoulos K, et al. Acute myeloid leukemia in a patient with Ataxia telangiectasia: A case report and review of literature. Leukemia 2001;15:1668-70.
- Taylor AM, Metcalfe JA, Thick J, Mak YF. Leukemia and lymphoma in Ataxia telangiectasia. Blood 1996;87:423-38.
- Yanofsky RA, Seshia SS, Dawson AJ, Stobart K, Greenberg CR, Booth FA, et al. Ataxia-telangiectasia: A typical presentation and toxicity of cancer treatment. Can J Neurol Sci 2009;36:462-7.
- Gungor T, Buhring I, Cremer R, Gartenschlagner M, Zeilen S. Pathogenesis, diagnosis, clinical and therapeutic aspects of ataxia telangiectasia. Klin Padiatr 1997;209:328-35.
| Figure 1:Case 1 (a) Peripheral smear showing leucocytosis with blast population; (b) Bone marrow showing predominantly of blast population; (c) Immunophenotyping showing positivity for T-lymphoid lineage markers (CD7, CD5) and early stem cell marker (CD34, human leucocyte antigen [HLA]-DR). CD4 and CD8 are negative
| Figure 2:Case 1 (a) Final karyotype report: 46, XY; (b) Chromosome rearrangement seen in two partial metaphases
| Figure 3:Family tree showing the relationship between the two cases
| Figure 4:Case 2 (a) Final karyotype report 46; (b): Chromosome rearrangement seen in two partial metaphases
References
- Huang KY, Shyur SD, Wang CY, Shen EY, Liang DC. Ataxia telangiectasia: Report of two cases. J Microbiol Immunol Infect 2001;34:71-5.
- Muller H. Recessively inherited deficiencies predisposing to cancer. Anticancer Res 1990;10:513-8.
- Kutukcluer N, Aksu G. Is there an association between autoimmune hemolytic anemia and ataxia-telangiectasia? Autoimmunity 2000;32:145-7.
- Folgori L, Scarselli A, Angelino G, Ferrari F, Antoccia A, Chessa L, et al. Cutaneous granulomatosis and combined immunodeficiency revealing Ataxia-Telangiectasia: A case report. Ital J Pediatr 2010;36:29.
- Loeb DM, Lederman HM, Winkelstein JA. Lymphoid malignancy as a presenting sign of ataxia-telangiectasia. J Paediatr Hematol Oncol 2000;22:464-7.
- Goyal V, Behari M. Dystonia as presenting manifestation of Ataxia Telangiectasia: A case report. Neurol India 2002;50:187-9.
- Staples ER, Mc Dermott EM, Reiman A, Byrd PJ, Ritchie S, Taylor AM, et al. Immunodeficiency in ataxia telangiectasia is correlated strongly with the presence of two null mutations in the ataxia telangiectasia mutated gene. Clin Exp Immunol 2008;153:214-20.
- Gatti RA. Ataxia Telangiectasia. Dermatol Clin 1995;13:1-6.
- Viniou N, Terpos E, Rombos J, Vaiopoulos G, Nodaras K, Stamatopoulos K, et al. Acute myeloid leukemia in a patient with Ataxia telangiectasia: A case report and review of literature. Leukemia 2001;15:1668-70.
- Taylor AM, Metcalfe JA, Thick J, Mak YF. Leukemia and lymphoma in Ataxia telangiectasia. Blood 1996;87:423-38.
- Yanofsky RA, Seshia SS, Dawson AJ, Stobart K, Greenberg CR, Booth FA, et al. Ataxia-telangiectasia: A typical presentation and toxicity of cancer treatment. Can J Neurol Sci 2009;36:462-7.
- Gungor T, Buhring I, Cremer R, Gartenschlagner M, Zeilen S. Pathogenesis, diagnosis, clinical and therapeutic aspects of ataxia telangiectasia. Klin Padiatr 1997;209:328-35.