 PDF
PDF  Views
Views  Share
Share


Clear cell sarcoma of kidney: A rare entity
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2011; 32(03): 157-159
DOI: DOI: 10.4103/0971-5851.92817
Abstract
Clear cell sarcoma of the kidney is an uncommon renal neoplasm of childhood. It represents one of the most common unfavorable tumors included in National Wilms′ Tumor Study Group clinical protocols. We came across this rare tumor in a 2-year-old male child. The case report is followed by discussion, stating the differentiating features between Wilms′ and clear cell sarcoma, histological details, treatment, and prognostication.
Keywords
Blastemal Wilms′ tumor - bone-metastasizing renal tumor of childhood - clear cell sarcoma of the kidneyPublication History
Article published online:
06 August 2021
© 2011. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Clear cell sarcoma of the kidney is an uncommon renal neoplasm of childhood. It represents one of the most common unfavorable tumors included in National Wilms’ Tumor Study Group clinical protocols. We came across this rare tumor in a 2-year-old male child. The case report is followed by discussion, stating the differentiating features between Wilms’ and clear cell sarcoma, histological details, treatment, and prognostication.
INTRODUCTION
Originally called “Bone-metastasizing renal tumor of childhood” by Marsden et al.,[1] clear cell sarcoma of the kidney (CCSK) is a highly malignant neoplasm occurring in the same age range as Wilms’ tumor. However, CCSK is resistant to conventional therapy, but often responsive to doxorubicin containing regimens.
CASE REPORT
A 2-year-old boy presented with 2-week history of a mass in the right upper abdomen. There was no history of hematuria and weight loss. Clinical examination showed firm, non-tender swelling occupying the right hypochondrium and lumbar region. Ultrasonography showed a 12.3 × 8.7 cm hetrogenous mass in the right kidney. With the provisional diagnosis of Wilms’ tumor it was removed en masse along with right ureter.
Gross examination revealed a right nephrectomy specimen, measuring 12.5 × 10.5 × 7.5 cm, encased in capsule surrounded by perinephric fat. Portion of lower pole of kidney was grossly identifiable, and there was a small segment of ureter measuring 0.75 cm located at the posteriomedial aspect along with vascular channels, embedded in the fat. Cut surface showed a circumscribed tumor mass measuring 9.5 × 9.0 × 7.0 cm. The surface was grayish solid with areas of necrosis and hemorrhage [Figure 1]. The advancing tumor border was identified in the lower pole only. Perinephric fat was grossly free from the tumor and the outer capsular surface appeared smooth. Adrenal gland was seen on the upper part of renal mass. A lymph node was also removed from the same area.

| Figure 1: Clear cell sarcoma of the kidney. The gray-tan tumor is large, soft focally cystic and necrotic
Sections showed highly cellular tumor consisting of proliferating monotonous undifferentiated malignant cells of small to medium size. Cell cytoplasm was moderate clear or faintly eosinophilic. Round to oval appearing nuclei showed anisokaryosis, with inconspicuous nucleoli. Mitosis was 1-5/hpf and atypical. Tumor showed delicate vascular stroma, foci of cystic degeneration, and extensive areas of necrosis [Figure 2]. Tumor infiltration was seen beyond the renal capsule with vascular embolization. Hilar node was negative for tumor cells. Immunochemical markers Vimentin, non specific esterase (NSE), and cytokeratin were used. Cytoplasmic focal expression for Vimentin was only detected.
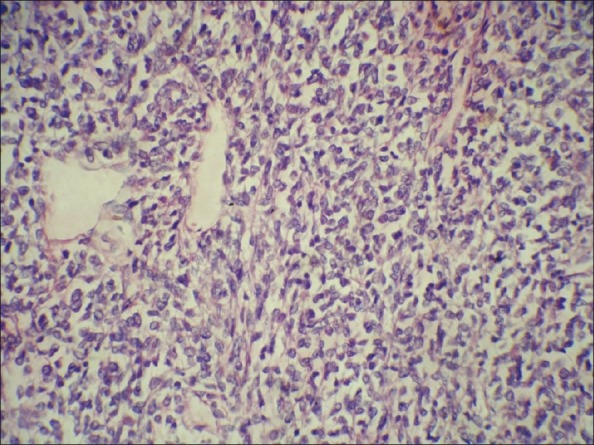
| Figure 2: Clear cell sarcoma of the kidney. The prominent and branching vascular patterns and cell nuclei with fine occasionally vesicular chromatin and inconspicuous nucleoli (H and E, ×HP)
In view of the above morphological and immunochistochemial findings, diagnosis of CCSK was confirmed. The bone scan was done prior to chemoradiation, which showed disseminated bony metastasis (stage 4). The patient was treated in accordance with NWTSG-3 protocol and was kept on doxorubicin in combination with vincristine and dactinomycin, initially for a period of 24 weeks. The patient also received postoperatively, a total of 10.8 Gy of radiation therapy in divided doses for the tumor bed. He responded to treatment as evident by clinical improvement at subsequent follow-ups till 4 months. The patient, however, died 2 months later at his home, and the cause of death remains unknown.
DISCUSSION
Non-Wilms’ renal tumors account for less than 10% of primary renal neoplasm of childhood.[2] Clear cell sarcoma also known as “Bone metastasizing tumor of Childhood” comprise 4% of all primary childhood renal tumors.[3] Some patients have a family history of this tumor. It is classified separately form Wilms’ tumor due to several distinctive features.[4,5] It has a peak incidence during 3-5 years, with a male:female ratio of 2:1.[6] CCSK is extremely rare in infants younger than 6 months and in young adults.
These are unilateral and unicentric tumors arising from the medullary region of the kidney with mucoid texture, foci of necrosis, and cyst formations. It is called clear cell because of the presence of numerous intracytoplasmic vesicles. CCSK has a propensity to permeate through the renal and perirenal vascular system.[7] Most of these have a classic pattern, With (91%)[4] as either a predominant or a secondary morphology.
The variant patterns recognized are myxoid (50%), sclerosing (35%), cellular (26%), epithelioid (trabecular or acinar type) (13%), palisading verocay body (11%), spindle cell (7%), storiform (4%), anaplastic (2.6%),[4] blended smoothly with classic pattern or with each other.
In the classic pattern, the tumor cells appear monomorphic with cords or nests of 6-10 cells separated by dispersed in small, regularly spaced arborizing fibrovascular septa. While usually plump, the cells in the core uncommonly may assume a spindle shape. Nuclei are overall uniform in shape, with fine dusty chromatin without prominent nucleoli or coarse condensations. Empty appearing Orphan Annie eye nuclei are a frequent occurrence. Cytoplasm is sparse and borders indistinct.[6]
Immunohistochemical staining show reactivity for Vimentin in the form of perinuclear dot like pattern.[8] Tumor cells are not reactive for Cytokeratin, EMA, MIC2, S100, Synaptophysin, GFAP, LCA, and NSE.
Management requires aggressive surgical approach followed by chemotherapy and radiotherapy as per NWTSG protocols. Relapses, although late, are common even in stage one tumors. The overall survival is 69%.[6]
Distinction of CCSK from blastemal Wilms’ tumor and primitive neuroectodermal tumor (PNET), both having prominent vascular patterns, is important. These tumors are more aggresively invasive than CCSK, entrapping whole islands of native renal parenchyma, as opposed to single tubules entrapped by CCSK. Wilms’ tumor has an early metastasis and classical spread to lymph nodes, lungs, and liver, but metastasis to bone is rare. Wilms’ tumor may be bilateral in 5% of the cases but CCSK is never bilateral. Nearly 10% of Wilms’ tumor displays recognized congenital anomalies such as overgrowth syndrome, isolated aniridia and trisomy 18, hypospadias, cryptorchidism, and renal fusion. CCSK has not shown any association with congenital anomalies.[9]
It is of considerable therapeutic importance that clear cell sarcoma be not only correctly diagnosed but early too.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
| Figure 1: Clear cell sarcoma of the kidney. The gray-tan tumor is large, soft focally cystic and necrotic
| Figure 2: Clear cell sarcoma of the kidney. The prominent and branching vascular patterns and cell nuclei with fine occasionally vesicular chromatin and inconspicuous nucleoli (H and E, ×HP)