 PDF
PDF  Views
Views  Share
Share


Clinicopathological Features and Treatment Outcomes in Ewing’s Sarcoma Patients: A 10 year experience of Alexandria Clinical Oncology Department
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2017; 38(03): 316-320
DOI: DOI: 10.4103/ijmpo.ijmpo_107_17
Abstract
Background: Ewing's sarcoma (ES) is an aggressive tumor, which is usually associated with micrometastases in the circulation. Thus, systemic chemotherapy in addition to local control modality is essential to improve outcomes. The aim of this study was to evaluate clinicopathological features and treatment outcomes in patients with ES. Materials and Methods: Medical files of 74 patients with nonmetastatic ES treated at our centers between 2004 and 2014 were retrospectively evaluated. The clinicopathological parameters were extracted and statistically correlated with event-free survival (EFS) and overall survival (OS). Results: The median age of patients was 13 years. The median follow-up duration was 63.8 months. About two-thirds (58.1%) of patients were male. Pain (74.3%) was the most common presenting symptom. Extremities (48.6%) were the frequently affected sites. Thirty-two patients (43.2%) presented by tumors larger than 8 cm. All patients were treated with chemotherapy. Local therapies were surgery and/or radiotherapy. The 5-year EFS and OS were 44% and 57%, respectively. On multivariate analysis, EFS and OS were significantly associated with age, tumor site, and tumor size. Conclusions: Despite limited resources in a developing country, the survival rates of ES are comparable to that in developed countries, and prognostic factors are age, tumor site, and tumor size.
Publication History
Article published online:
04 July 2021
© 2017. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Background:
Ewing's sarcoma (ES) is an aggressive tumor, which is usually associated with micrometastases in the circulation. Thus, systemic chemotherapy in addition to local control modality is essential to improve outcomes. The aim of this study was to evaluate clinicopathological features and treatment outcomes in patients with ES.
Materials and Methods:
Medical files of 74 patients with nonmetastatic ES treated at our centers between 2004 and 2014 were retrospectively evaluated. The clinicopathological parameters were extracted and statistically correlated with event-free survival (EFS) and overall survival (OS).
Results:
The median age of patients was 13 years. The median follow-up duration was 63.8 months. About two-thirds (58.1%) of patients were male. Pain (74.3%) was the most common presenting symptom. Extremities (48.6%) were the frequently affected sites. Thirty-two patients (43.2%) presented by tumors larger than 8 cm. All patients were treated with chemotherapy. Local therapies were surgery and/or radiotherapy. The 5-year EFS and OS were 44% and 57%, respectively. On multivariate analysis, EFS and OS were significantly associated with age, tumor site, and tumor size.
Conclusions:
Despite limited resources in a developing country, the survival rates of ES are comparable to that in developed countries, and prognostic factors are age, tumor site, and tumor size.
Introduction
Ewing's sarcoma (ES) family is a group of malignant small round cell tumors of neuroectodermal origin that vary in their neurogenic differentiation but share the same treatment and prognosis. ES is the second most frequent primary malignant bone tumor that affects mainly children and young adult and constitutes 3% of all pediatric malignancies.[1] It rarely occurs in adults over the age of 30.[1]
The survival rates in patients with ES have been markedly improved due to the advent of recent chemotherapy protocols, advanced radiotherapeutic techniques with accurate localization and delivery, and improved surgical techniques.[2,3,4,5,6] The prognosis of patients with ES depends on the age of the patient, stage of disease, primary tumor site, and tumor size.[7,8,9,10]
Despite growing literature evaluating the treatment effectiveness in patients with ES from the Western countries, data assessing outcomes of currently utilized therapy from the Middle East are scarce. Furthermore, conflicting results have been reported.[2,3,4,5,6,11,12] Thus, this study was designed to identify the clinicopathological features and treatment results of patients presented to our tertiary centers.
Materials and Methods
The medical records of patients with histologically confirmed ES presented to Alexandria Main University Hospital and Alexandria Sporting Students' Hospital during the period from January 2004 to December 2014 were reviewed. Patients with metastatic disease at presentation or had incomplete information were excluded from the study. A total of 74 patients were included with a median follow-up duration of 63.8 months. Data regarding age at diagnosis, sex, site and size (based on the greatest reported dimension) of the primary tumor, chemotherapy, surgery, radiotherapy, relapse, and survivals were extracted.
Statistical analysis
Data were analyzed using SPSS software version 22 (IBM Corp. Released 2013. IBM SPSS Statistics for Windows, Version 22.0. Armonk, NY: IBM Corp.) (http://www.ibm.com). Overall survival (OS) was calculated from the date of diagnosis to the date of death or date of the last follow-up. Event-free survival (EFS) was calculated from the date of diagnosis to the date of occurrence of an adverse event (disease progression, second malignant neoplasm, or death) or date of the last follow-up, whichever came first. Disease progression was further classified as local progression (recurrence at the initial site only), systemic progression (recurrence at a site not initially involved by disease), or local plus systemic progression (at the initial site and another site). Patients without disease recurrence were censored at date of the last follow-up.
Age was considered as a categorical variable (<10>15 years) using age group 10–15 years as a reference. Other clinical parameters of sex (male vs. female), primary tumor site (extremities vs. pelvis vs. others), tumor size (≤8 cm vs. >8 cm), and local control modality (radiotherapy vs. surgery vs. surgery + radiotherapy) were considered as categorical variables using the first subset as an indicator. OS and EFS curves were calculated using Kaplan–Meier method and compared using the log-rank test in univariate analysis. The relative influence of different prognostic factors on survival was estimated with the Cox regression model. P ≤ 0.05 was considered statistically significant.
Results
Patients' characteristics and treatment
Table 1 summarizes the characteristics of patients. The median age of patients was 13 years (range: 4–22 years). About two-thirds (58.1%) of patients were male. Pain was the most common presenting symptom and occurred in 55 patients (74.3%) followed by swelling in 45 patients (60.8%). Extremities were the frequently affected sites (48.6% of patients) followed by pelvis (29.7% of patients). Thirty-two patients (43.2%) presented by tumors larger than 8 cm. All patients received uniform chemotherapy (cycles of vincristine-doxorubicin-cyclophosphamide alternated with cycles of ifosfamide-etoposide and repeated every 3 weeks). Regarding local control modality, 33 patients (44.6%) were treated with radiotherapy alone, 22 patients (29.7%) underwent surgery alone, and 19 patients (25.7%) underwent both surgery and radiotherapy.
Table 1
Patients and disease characteristics

|
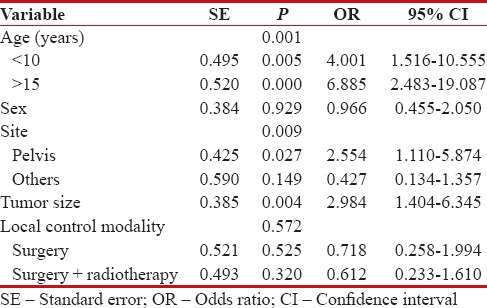
The 5-year EFS of patients was 44%. On univariate analysis [Figure 1], EFS was not significantly affected by gender (P = 0.625). However, EFS was significantly associated with age (P = 0.00), tumor site (P = 0.00), tumor size (P = 0.005), and local control modality (P = 0.007). On multivariate analysis [Table 2], EFS was significantly associated with age (P = 0.001), tumor site (P = 0.009), and tumor size (P = 0.004).
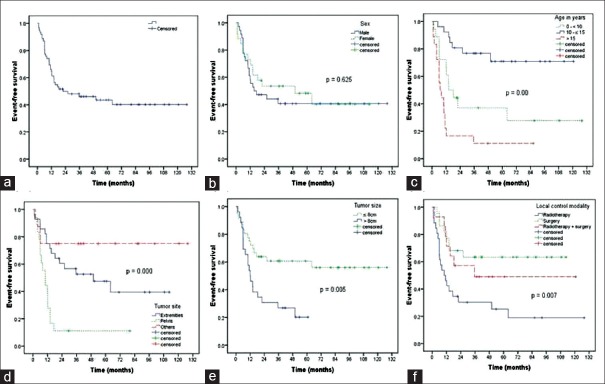
| Figure 1:Kaplan–Meier estimate of event-free survival for 74 patients with nonmetastatic Ewing's sarcoma (a) and shown according to sex (b), age (c), tumor site (d), tumor size (e), and local control modality (f)
Table 2
Multivariate analysis for event-free survival of 74 patients with nonmetastatic Ewing's sarcoma
|
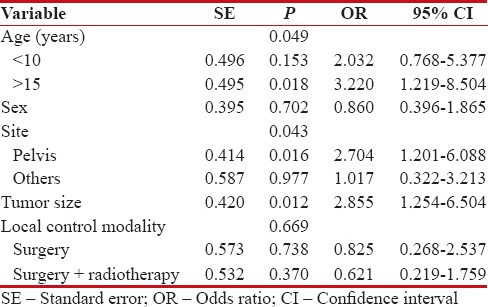
The 5-year OS of patients was 57%. On univariate analysis [Figure 2], age (P = 0.001), tumor site (P = 0.00), tumor size (P = 0.001), and local control modality (P = 0.013) were significantly correlated to OS. However, in multivariate analysis [Table 3], age (P = 0.049), tumor site (P = 0.043), and tumor size (P = 0.012) were significantly associated with OS.

| Figure 2:Kaplan–Meier estimate of overall survival for 74 patients with nonmetastatic Ewing's sarcoma (a) and shown according to sex (b), age (c), tumor site (d), tumor size (e), and local control modality (f)
Table 3
Multivariate analysis for overall survival of 74 patients with nonmetastatic Ewing's sarcoma
|
Discussion
ES is a very aggressive disease, and multimodality treatment approach that involves local control modality with attempts to preserve the function, and eradication of micrometastases using chemotherapy is the current standard treatment of ES.[2,3,4,5,6,7] We tried in this work to determine the clinicopathological features and treatment outcomes of patients with nonmetastatic ES in an Egyptian population.
The median age of our patients was 13 years. Consistently, the median age was 13.7 years in the review of St. Jude Children's Research Hospital studies carried out by Rodríguez-Galindo et al.[7] Other studies by Biswas et al. and Paulussen et al. reported that the median age was 15 years which is not significantly different from ours.[8,13]
In this study, 58% of patients were male. Male predominance was also seen in other studies. Rodríguez-Galindo et al. reported that 60.9% of ES were male.[7] Paulussen et al. analyzed 301 patients and found that 60% of patients were male.[13] Akhavan et al. evaluated 32 patients with ES and reported 65.2% were male.[12] Jurgens et al., Obata et al., and Haeusler et al. found the same results.[2,4,14]
Pain was the most common presenting symptom in our patients (74.3%). This is similar to Akhavan et al., who reported that pain was the first symptom in 83.8% of patients with ES.[12] In another study, the authors evaluated 47 patients with ES and concluded that the initial symptom was pain.[15]
In our study, extremities were the frequently affected sites (48.6% of patients). This agrees with literature. Extremities were the primary site of disease in 47%, 46%, and 31.6% of patient in studies performed by Obata et al., Oksüz et al., and Haeusler et al., respectively.[2,14,16]
In the current study, the 5-year EFS for patients with nonmetastatic ES was 44%. Our results are consistent with the results of the Japanese study carried out by Obata et al., who reported that the 5-year disease-free survival rates in nonmetastatic patients with ES were 46.6%.[14] Oksüz et al. retrospectively evaluated 65 patients with nonmetastatic ES of the bone and reported that the 5-year EFS was 44%.[16]
In this study, the 5-year OS for patients with nonmetastatic ES was 57%. This is similar to the results obtained in other studies. Biswas et al. reviewed 158 patients and reported that the 5-year OS was 57.6% for nonmetastatic ES patients.[8] Obata et al. in the Japanese study found that the 5-year OS in nonmetastatic ES patients was 54.9%.[14] Lee et al. demonstrated that the 5-year OS for nonmetastatic ES patients was 61.6%.[17]
In the present study, the most important prognostic factors for both EFS and OS for nonmetastatic ES were age at diagnosis, tumor site, and tumor size. Older patients with age >15 years, patients with tumor size ≥8 cm, or patients with pelvic primary tumor had significantly poor EFS and OS in our study. Consistent with our results, Oksüz et al. found in a multivariate analysis that age, tumor site, and tumor size were the prognostic factors for ES patients.[16] The authors reported that both EFS and OS were worse for patients >17 years of age, tumor size >8 cm in diameter, or an axial location.[16] In the Japanese study, Obata et al. demonstrated that patients who had primary site in the trunk and age ≥16 years had a significantly worse prognosis.[14]
Consistent with our results, studies have showed that tumor size has an important effect on the outcome.[7,9,10] Paulussen et al. analyzed 301 patients with ES and found in multivariate analysis that sizable tumor volume had negative impacts on EFS.[13] Both Tural et al. and Arpaci et al. reported that patients with tumor size ≥8 cm had significantly worse EFS and OS.[9,10] Large tumors are associated with worse outcomes, as large tumors are usually irresectable and associated with micrometastases. Large tumors are more radioresistant due to increased hypoxia and necrosis. Moreover, the number of cancer stem cells requiring sterilization increases in direct proportion with tumor size.[18]
Our results are supported by literature where several reports have suggested that patients with pelvic ES had an unfavorable prognosis.[19,20,21] This might be due to difficulties in the surgical approach of the tumor, sizable tumor at presentation, or the intrinsic behavior of the pelvic tumor. In a Finnish nationwide study on bone and soft tissue ES, Serlo et al. found that young age at diagnosis and a peripheral primary tumor site were associated with a better prognosis.[21] Donaldson et al. found that site of primary tumor correlated with 5-year EFS.[19] Cotterill et al. demonstrated in multivariate analysis that tumor site and age group (<15 href="https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5686974/#ref20" rid="ref20" class=" bibr popnode tag_hotlink tag_tooltip" id="__tag_649899141" role="button" aria-expanded="false" aria-haspopup="true" xss=removed>20]
To our knowledge, this is the first study on ES treated with uniform chemotherapy and either surgical resection and/or radiation therapy with the longest follow-up period from Egypt.
Despite genetic differences and limited resources, it seems that the clinical features, prognostic factors, and treatment outcome of ES patients in our centers are comparable to the outcomes observed in other parts of the world. This is because of the use of multidisciplinary approach and following international protocols.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Siegel DA, King J, Tai E, Buchanan N, Ajani UA, Li J. Cancer incidence rates and trends among children and adolescents in the United States, 2001-2009. Pediatrics 2014;134:e945-55.
- Haeusler J, Ranft A, Boelling T, Gosheger G, Braun-Munzinger G, Vieth V, et al. The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 2010;116:443-50.
- Ginsberg JP, Goodman P, Leisenring W, Ness KK, Meyers PA, Wolden SL, et al. Long-term survivors of childhood Ewing sarcoma: Report from the childhood cancer survivor study. J Natl Cancer Inst 2010;102:1272-83.
- Jürgens H, Exner U, Gadner H, Harms D, Michaelis J, Sauer R, et al. Multidisciplinary treatment of primary Ewing's sarcoma of bone. A 6-year experience of a European Cooperative Trial. Cancer 1988;61:23-32.
- Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003;348:694-701.
- Burgert EO Jr. Nesbit ME, Garnsey LA, Gehan EA, Herrmann J, Vietti TJ, et al. Multimodal therapy for the management of nonpelvic, localized Ewing's sarcoma of bone: Intergroup study IESS-II. J Clin Oncol 1990;8:1514-24.
- Rodríguez-Galindo C, Liu T, Krasin MJ, Wu J, Billups CA, Daw NC, et al. Analysis of prognostic factors in Ewing sarcoma family of tumors: Review of St. Jude Children's Research Hospital Studies. Cancer 2007;110:375-84.
- Biswas B, Rastogi S, Khan SA, Mohanti BK, Sharma DN, Sharma MC, et al. Outcomes and prognostic factors for Ewing-family tumors of the extremities. J Bone Joint Surg Am 2014;96:841-9.
- Tural D, Molinas Mandel N, Dervisoglu S, Oner Dincbas F, Koca S, Colpan Oksuz D, et al. Extraskeletal Ewing's sarcoma family of tumors in adults: Prognostic factors and clinical outcome. Jpn J Clin Oncol 2012;42:420-6.
- Arpaci E, Yetisyigit T, Seker M, Uncu D, Uyeturk U, Oksuzoglu B, et al. Prognostic factors and clinical outcome of patients with Ewing's sarcoma family of tumors in adults: Multicentric study of the Anatolian Society of Medical Oncology. Med Oncol 2013;30:469.
- Abou Ali B, Nader R, Tamim H, Eid T, Boulos F, Khoury N, et al. Outcome of Ewing sarcoma in a multidisciplinary setting in Lebanon. Pediatr Blood Cancer 2014;61:1472-5.
- Akhavan A, Binesh F, Shamshiri H, Ghanadi F. Survival of patients with Ewing's sarcoma in Yazd-Iran. Asian Pac J Cancer Prev 2014;15:4861-4.
- Paulussen M, Ahrens S, Dunst J, Winkelmann W, Exner GU, Kotz R, et al. Localized Ewing tumor of bone: Final results of the cooperative Ewing's Sarcoma Study CESS 86. J Clin Oncol 2001;19:1818-29.
- Obata H, Ueda T, Kawai A, Ishii T, Ozaki T, Abe S, et al. Clinical outcome of patients with Ewing sarcoma family of tumors of bone in Japan: The Japanese Musculoskeletal Oncology Group cooperative study. Cancer 2007;109:767-75.
- Widhe B, Widhe T. Initial symptoms and clinical features in osteosarcoma and Ewing sarcoma. J Bone Joint Surg Am 2000;82:667-74.
- Oksüz DC, Tural D, Dincbas FÖ, Dervisoglu S, Turna H, Hiz M, et al. Non-metastatic Ewing's sarcoma family of tumors of bone in adolescents and adults: Prognostic factors and clinical outcome-single institution results. Tumori 2014;100:452-8.
- Lee CY, Yen CC, Yen HJ, Shiau CY, Chao TC, Wu PK, et al. Outcomes of 50 patients with Ewing sarcoma family of tumors treated at a single institution in Taiwan. Medicine (Baltimore) 2016;95:e3830.
- ;Bentzen SM, Thames HD. Tumor volume and local control probability: Clinical data and radiobiological interpretations. Int J Radiat Oncol Biol Phys 1996;36:247-51.
- Donaldson SS, Torrey M, Link MP, Glicksman A, Gilula L, Laurie F, et al. A multidisciplinary study investigating radiotherapy in Ewing's sarcoma: End results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 1998;42:125-35.
- Cotterill SJ, Ahrens S, Paulussen M, Jürgens HF, Voûte PA, Gadner H, et al. Prognostic factors in Ewing's tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol 2000;18:3108-14.
- Serlo JA, Helenius IJ, Sampo M, Vettenranta K, Saarinen-Pihkala UM, Kivivuori SM, et al. Ewing's sarcoma family of tumors in Finland during 1990-2009: A population-based study. Acta Oncol 2013;52:767-75.
| Figure 1:Kaplan–Meier estimate of event-free survival for 74 patients with nonmetastatic Ewing's sarcoma (a) and shown according to sex (b), age (c), tumor site (d), tumor size (e), and local control modality (f)
| Figure 2:Kaplan–Meier estimate of overall survival for 74 patients with nonmetastatic Ewing's sarcoma (a) and shown according to sex (b), age (c), tumor site (d), tumor size (e), and local control modality (f)
References
- Siegel DA, King J, Tai E, Buchanan N, Ajani UA, Li J. Cancer incidence rates and trends among children and adolescents in the United States, 2001-2009. Pediatrics 2014;134:e945-55.
- Haeusler J, Ranft A, Boelling T, Gosheger G, Braun-Munzinger G, Vieth V, et al. The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 2010;116:443-50.
- Ginsberg JP, Goodman P, Leisenring W, Ness KK, Meyers PA, Wolden SL, et al. Long-term survivors of childhood Ewing sarcoma: Report from the childhood cancer survivor study. J Natl Cancer Inst 2010;102:1272-83.
- Jürgens H, Exner U, Gadner H, Harms D, Michaelis J, Sauer R, et al. Multidisciplinary treatment of primary Ewing's sarcoma of bone. A 6-year experience of a European Cooperative Trial. Cancer 1988;61:23-32.
- Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003;348:694-701.
- Burgert EO Jr. Nesbit ME, Garnsey LA, Gehan EA, Herrmann J, Vietti TJ, et al. Multimodal therapy for the management of nonpelvic, localized Ewing's sarcoma of bone: Intergroup study IESS-II. J Clin Oncol 1990;8:1514-24.
- Rodríguez-Galindo C, Liu T, Krasin MJ, Wu J, Billups CA, Daw NC, et al. Analysis of prognostic factors in Ewing sarcoma family of tumors: Review of St. Jude Children's Research Hospital Studies. Cancer 2007;110:375-84.
- Biswas B, Rastogi S, Khan SA, Mohanti BK, Sharma DN, Sharma MC, et al. Outcomes and prognostic factors for Ewing-family tumors of the extremities. J Bone Joint Surg Am 2014;96:841-9.
- Tural D, Molinas Mandel N, Dervisoglu S, Oner Dincbas F, Koca S, Colpan Oksuz D, et al. Extraskeletal Ewing's sarcoma family of tumors in adults: Prognostic factors and clinical outcome. Jpn J Clin Oncol 2012;42:420-6.
- Arpaci E, Yetisyigit T, Seker M, Uncu D, Uyeturk U, Oksuzoglu B, et al. Prognostic factors and clinical outcome of patients with Ewing's sarcoma family of tumors in adults: Multicentric study of the Anatolian Society of Medical Oncology. Med Oncol 2013;30:469.
- Abou Ali B, Nader R, Tamim H, Eid T, Boulos F, Khoury N, et al. Outcome of Ewing sarcoma in a multidisciplinary setting in Lebanon. Pediatr Blood Cancer 2014;61:1472-5.
- Akhavan A, Binesh F, Shamshiri H, Ghanadi F. Survival of patients with Ewing's sarcoma in Yazd-Iran. Asian Pac J Cancer Prev 2014;15:4861-4.
- Paulussen M, Ahrens S, Dunst J, Winkelmann W, Exner GU, Kotz R, et al. Localized Ewing tumor of bone: Final results of the cooperative Ewing's Sarcoma Study CESS 86. J Clin Oncol 2001;19:1818-29.
- Obata H, Ueda T, Kawai A, Ishii T, Ozaki T, Abe S, et al. Clinical outcome of patients with Ewing sarcoma family of tumors of bone in Japan: The Japanese Musculoskeletal Oncology Group cooperative study. Cancer 2007;109:767-75.
- Widhe B, Widhe T. Initial symptoms and clinical features in osteosarcoma and Ewing sarcoma. J Bone Joint Surg Am 2000;82:667-74.
- Oksüz DC, Tural D, Dincbas FÖ, Dervisoglu S, Turna H, Hiz M, et al. Non-metastatic Ewing's sarcoma family of tumors of bone in adolescents and adults: Prognostic factors and clinical outcome-single institution results. Tumori 2014;100:452-8.
- Lee CY, Yen CC, Yen HJ, Shiau CY, Chao TC, Wu PK, et al. Outcomes of 50 patients with Ewing sarcoma family of tumors treated at a single institution in Taiwan. Medicine (Baltimore) 2016;95:e3830.
- ;Bentzen SM, Thames HD. Tumor volume and local control probability: Clinical data and radiobiological interpretations. Int J Radiat Oncol Biol Phys 1996;36:247-51.
- Donaldson SS, Torrey M, Link MP, Glicksman A, Gilula L, Laurie F, et al. A multidisciplinary study investigating radiotherapy in Ewing's sarcoma: End results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 1998;42:125-35.
- Cotterill SJ, Ahrens S, Paulussen M, Jürgens HF, Voûte PA, Gadner H, et al. Prognostic factors in Ewing's tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol 2000;18:3108-14.
- Serlo JA, Helenius IJ, Sampo M, Vettenranta K, Saarinen-Pihkala UM, Kivivuori SM, et al. Ewing's sarcoma family of tumors in Finland during 1990-2009: A population-based study. Acta Oncol 2013;52:767-75.