 PDF
PDF  Views
Views  Share
Share


Efficacy and Safety of Deferasirox in Pediatric Patients of Thalassemia at a Tertiary Care Teaching Hospital
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2017; 38(02): 103-110
DOI: DOI: 10.4103/ijmpo.ijmpo_103_16
Abstract
Objective: To evaluate efficacy, safety and utilization pattern of deferasirox in paediatric patients of transfusion dependant β Thalassemia Major at a tertiary care teaching hospital in Gujarat. Materials and Methods: This observational, prospective-retrospective, single centre, continuous study was conducted in a tertiary care teaching hospital among paediatric patients of transfusion dependent β Thalassemia Major. Patients treated with deferasirox for not more than 12 weeks were enrolled. Details of blood transfusions, relevant investigations performed every 3 weeks and 3 months and drugs used were recorded in a pretested case record form. Parents were provided with a diary to record the details of ADRs. Data were analyzed for demographic characteristics, number and mean volume of blood transfusions, changes in serum ferritin and iron levels, number and types of ADRs and progression, causality, severity and preventability of ADRs. Results: Of the 60 patients enrolled, one patient was lost to follow up and four withdrew their consent. Of the remaining 55 patients, 36 were boys and 19 were girls (mean age: 6 ± 3.14 years), including patients of 1-3 years (11), 4-6 years (24), 7-10 years (12) and 11-12 years (8). Thirty six patients were born of consanguineous marriages. Adherence to blood transfusion guidelines and deferasirox prescribing and administration guidelines was observed. A serial and significant decrease in mean serum ferritin and serum iron at 3 weeks and 3 months with deferasirox treatment was observed in all age groups except that of 11-12 years. A total of 117 ADRs were observed in 52 patients from 19498 doses, most common being diarrhea (24), raised serum creatinine (15), raised hepatic enzymes (14), abdominal pain (14) and rashes (14). A reduction in dose was required in 32 cases, while a temporary stoppage was indicated in 41 cases. Deferasirox was the possible and probable cause of 65 and 51 ADRs respectively as assessed by WHO-UMC scale. Majority of ADRs were definitely preventable and mild in nature. Conclusion: β Thalassemia Major is more common in males. A rational prescribing of deferasirox was observed. Deferasirox effectively reduced serum ferritin and serum iron levels in these patients.
Publication History
Article published online:
06 July 2021
© 2017. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Objective:
To evaluate efficacy, safety and utilization pattern of deferasirox in paediatric patients of transfusion dependant β Thalassemia Major at a tertiary care teaching hospital in Gujarat.
Materials and Methods:
This observational, prospective-retrospective, single centre, continuous study was conducted in a tertiary care teaching hospital among paediatric patients of transfusion dependent β Thalassemia Major. Patients treated with deferasirox for not more than 12 weeks were enrolled. Details of blood transfusions, relevant investigations performed every 3 weeks and 3 months and drugs used were recorded in a pretested case record form. Parents were provided with a diary to record the details of ADRs. Data were analyzed for demographic characteristics, number and mean volume of blood transfusions, changes in serum ferritin and iron levels, number and types of ADRs and progression, causality, severity and preventability of ADRs.
Results:
Of the 60 patients enrolled, one patient was lost to follow up and four withdrew their consent. Of the remaining 55 patients, 36 were boys and 19 were girls (mean age: 6 ± 3.14 years), including patients of 1-3 years (11), 4-6 years (24), 7-10 years (12) and 11-12 years (8). Thirty six patients were born of consanguineous marriages. Adherence to blood transfusion guidelines and deferasirox prescribing and administration guidelines was observed. A serial and significant decrease in mean serum ferritin and serum iron at 3 weeks and 3 months with deferasirox treatment was observed in all age groups except that of 11-12 years. A total of 117 ADRs were observed in 52 patients from 19498 doses, most common being diarrhea (24), raised serum creatinine (15), raised hepatic enzymes (14), abdominal pain (14) and rashes (14). A reduction in dose was required in 32 cases, while a temporary stoppage was indicated in 41 cases. Deferasirox was the possible and probable cause of 65 and 51 ADRs respectively as assessed by WHO-UMC scale. Majority of ADRs were definitely preventable and mild in nature.
Conclusion:
β Thalassemia Major is more common in males. A rational prescribing of deferasirox was observed. Deferasirox effectively reduced serum ferritin and serum iron levels in these patients.
Introduction
Thalassemia is a heterogeneous group of autosomal recessive disorders, leading to a reduced synthesis of globin chains required for hemoglobin synthesis. Clinical manifestations depend on the extent to which the synthesis of the affected globin chain is impaired, altered synthesis of other globin chains, and coinheritance of other abnormal globin alleles.[1]
An estimated 80–90 million people worldwide carry the β thalassemia trait.[2] β thalassemia is the most common single gene disorder in India,[3] with nearly 36 million people carrying the responsible gene.[4] Thalassemia major is the severe form of β thalassemia characterized by severe anemia, hepatosplenomegaly, and facioskeletal changes due to increased hemolysis of defective red blood cells (RBCs). Blood transfusion (BT) remains the first-line treatment in these patients. It improves anemia and suppresses ineffective erythropoiesis. One unit of blood contains approximately 200–250 mg of elemental iron,[5] and it can cause iron overload when transfused repeatedly. The excess iron gets deposited in various tissues of body. Transfusion-related iron overload has been associated with various complications, for example, growth retardation, endocrinal abnormalities, and cardiac failure.[6] To prevent complications of iron overload, iron chelating therapy is recommended for these patients.
Iron chelators remove excess of iron from the body by forming nontoxic, stable, and water soluble complexes. Desferrioxamine and deferiprone, the conventional iron chelators, require frequent administration due to their short plasma half-lives. Deferasirox is a new iron chelator, which requires once a day oral administration. However, limited data regarding efficacy and safety of this drug in Indian population are available. Hence, the present study evaluates the efficacy, safety, utilization pattern, and tolerability of deferasirox in pediatric patients with transfusion-dependent β thalassemia major. The information obtained from the study could prove useful to recommend modifications, if any, in the management of iron overload in these patients.
Subjects and Methods
This was a single-center observational, prospective-retrospective, continuous study carried out at the thalassemia clinic and pediatric wards of a tertiary care hospital. Approval for conduct of the study was obtained from the Institutional Ethics Committee (Reg. No.: EC/Approval/40/12) and the Head of Department of Paediatrics. Transfusion-dependent pediatric patients of β thalassemia major, who were prescribed deferasirox for not more than past 3 months, were enrolled over a 9-month period. Patients with nontransfusion hemosiderosis, those whose parents/guardians did not consent to participate, and patients who did not assent for the study were excluded from the study. The enrolled patients were evaluated every 3 weeks for 1 year. Relevant laboratory investigations were carried out at 3 weeks and 3 monthly thereafter. Efficacy of deferasirox was evaluated by analyzing changes in serum ferritin and serum iron levels. Safety and tolerability of the drug were evaluated by monitoring of adverse drug reactions (ADRs) including changes in complete blood count, renal function tests, and liver function tests. In addition, parents were provided a diary to record any ADR observed by them. Data of each patient were recorded in a pretested case record form and analyzed for demographic characteristics, details of BTs, and changes in serum ferritin and serum iron levels. The number, type, progression, causality, severity, and preventability of observed ADRs were also evaluated. Causality of ADRs was assessed using WHO-Uppsala Monitoring Centre (UMC) Scale and Naranjo's algorithm. Severity of ADRs was assessed by modified Hartwig and Siegel scale. Modified Schumock and Thorton criteria were used to assess preventability of ADRs. Paired Student's t-test, unpaired Student's t-test, Pearson's R-test, and one-way ANOVA test were used for statistical analysis. IBM SPSS Statistics (version 20, IBM Corp., NY) and GraphPad InStat (version 3.10, San Diego, U.S.A.) were used for statistical analysis.
Results
A total 10,980 pediatric patients were admitted annually to this tertiary care hospital. Of these, 44 patients were newly diagnosed to suffer from β thalassemia major, accounting for an incidence of approximately 4% of β thalassemia major in this population. A total of sixty patients were enrolled. One patient was lost to follow-up, while four patients withdrew their consent from the study. Relevant data of remaining 55 patients (accounting for 330 follow-up visits) were recorded, which included retrospective data of 3 months from 15 patients.
Of the 55 patients, 36 were males and 19 were females (male:female ratio of 1.89:1). Mean age of these patients was 6.0 ± 3.14 years and most patients (n = 24, 43.6%) belonged to the age group of 4–6 years [Figure 1]. Of the 55 patients, majority (n = 40) belonged to Devipujak (n = 13), Muslim (n = 9), Sindhi (n = 6), Chamar (n = 6), and Kshatriya (n = 6) communities. Thirty-six patients (65.4%) were born of consanguineous marriage. Majority of the patients (n = 46, 83.6%) were diagnosed with β thalassemia major between the age of 6 and 12 months; the mean age at diagnosis was 14.05 ± 9.8 months. Left ventricular ejection fraction (LVEF) was normal in 46 patients at baseline as well as at the end of the study. Low LVEF was detected in nine cases (16.3%) at baseline (mean LVEF of 49.8% ± 2.08%). A significant improvement in mean LVEF as compared to baseline was observed in these cases at the end of the study (mean LVEF of 50.77% ± 2.22%; P < 0>
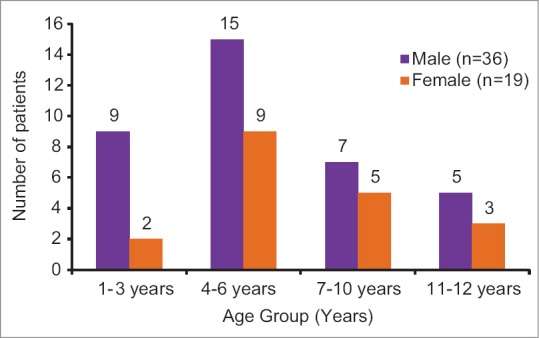
| Figure 1Age and gender distribution of children with β thalassemia major reporting at a tertiary care teaching hospital (n = 55)
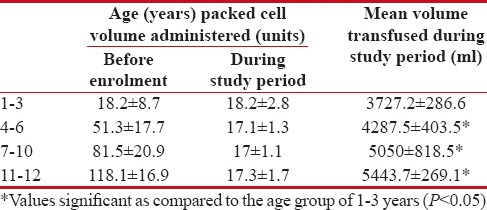
All the enrolled patients received packed cell volume (PCV) BTs. The patients received a total of 956 BTs during the study period (an average of 1.93 BTs per patient per month). The mean volume of blood transfused was significantly lower in patients of all age group of 1–3 years as compared to other age groups (P < 0 href="https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5582544/table/T1/" target="table" class="fig-table-link figpopup" rid-figpopup="T1" rid-ob="ob-T1" co-legend-rid="" xss=removed>Table 1]. Adherence to BT guidelines (Indian Academy of Paediatrics [IAP], 2006) was observed in both splenectomized (n = 9) (mean volume of 4444.4 ± 1210.7 ml; an average of 131.8 ml/kg/year) and nonsplenectomized patients (n = 46) (mean volume of 4345.4 ± 1673.4 ml; an average of 180.3 ml/kg/year).
Table 1
Mean number and volume of blood transfused in children with β-thalassemia major at a tertiary care teaching hospital (n=55)
|
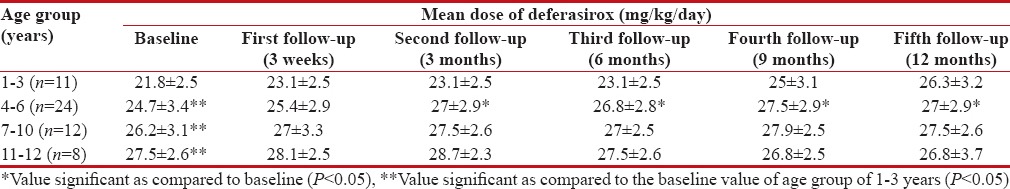
It was observed that all patients (baseline mean serum ferritin level of 2818 ± 879.6 ng/ml) were prescribed deferasirox, once a day in varying doses (20, 25 or 30 mg/kg/day); the dose was determined based on the serum ferritin levels. A total of 19,498 doses of deferasirox were administered orally over an average duration of 11.7 months during the entire study period. Deferasirox was prescribed by brand name “Asunra®” and dispensed as dispersible tablets and supplied free of cost from the hospital pharmacy. The IAP guidelines (IAP, 2006) for administration of deferasirox were adhered to in a majority of patients (n = 40, 72.7%). However, these were not complied within twenty incidents in fifteen patients. In these cases, the drug was administered either with food (n = 14) or without proper dispersion in water or orange juice (n = 6). A change in dose of deferasirox was required in 57 ADRs. This included 25 cases, where dose was increased to improve iron chelation, while in 32 cases, a reduction in the dose was required due to occurrence of ADRs. Drug was temporarily stopped in 41 cases due to raised serum creatinine or liver enzymes. Mean baseline dose of deferasirox was lower in patients of age group of 1–3 years as compared to other age groups (P < 0 xss=removed>P < 0 xss=removed>P < 0 xss=removed>P < 0 href="https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5582544/table/T2/" target="table" class="fig-table-link figpopup" rid-figpopup="T2" rid-ob="ob-T2" co-legend-rid="" xss=removed>Table 2].
Table 2
Mean prescribed dose of deferasirox in children with β thalassemia major at tertiary care teaching hospital (n=55)
|
Serum ferritin levels were analyzed in 52 patients (baseline data of three patients were not available). The baseline mean serum ferritin levels were significantly higher in age groups of 7–10 years and 11–12 years as compared to the age group of 1–3 years (P < 0 xss=removed>P < 0 href="https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5582544/table/T3/" target="table" class="fig-table-link figpopup" rid-figpopup="T3" rid-ob="ob-T3" co-legend-rid="" xss=removed>Table 3]. A significant decrease in serum ferritin levels was observed in all age groups at the end of 1 year of treatment with deferasirox.
Table 3
Mean serum ferritin in children with β thalassemia major following treatment with deferasirox (n=55)

|
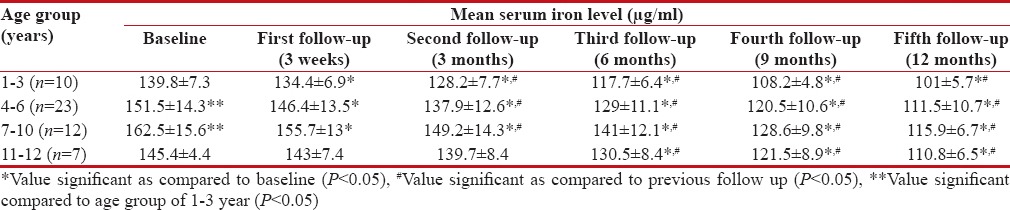
Serum iron levels were analyzed in 52 patients (three patients were excluded as described earlier). The baseline mean serum iron level was higher in patients of age groups of 4–6 years and 7–10 years as compared to age group of 1–3 years (P < 0 xss=removed>P < 0 href="https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5582544/table/T4/" target="table" class="fig-table-link figpopup" rid-figpopup="T4" rid-ob="ob-T4" co-legend-rid="" xss=removed>Table 4].
Table 4
Mean serum iron in children with β thalassemia major following treatment with deferasirox (n=55)
|
A significant negative correlation was observed between mean dose of deferasirox and mean reduction in serum ferritin (r = −0.941, P = 0.005) and between mean dose of deferasirox and mean serum iron (r = −0.931, P = 0.006) in age group of 1–3 years. Similarly, a significant negative correlation was observed between mean dose of deferasirox and mean serum iron (r = −0.835, P = 0.03) in age group of 4–6 years. No significant correlation was observed between mean dose of deferasirox and mean serum ferritin in age groups of 4–6 years (r = −0.770, P = 0.07) and 7–10 years (r = −0.718, P = 0.1). No significant correlation was observed between mean dose of deferasirox and mean serum iron in age group of 7–10 years (r = −0.738, P = 0.09). In the age group of 11–12 years, a positive but nonsignificant correlation was observed between mean dose of deferasirox and mean serum ferritin (r = 0.746, P = 0.08) and the mean dose of deferasirox and mean serum iron (r = 0.756, P = 0.08).
Adverse drug reactions
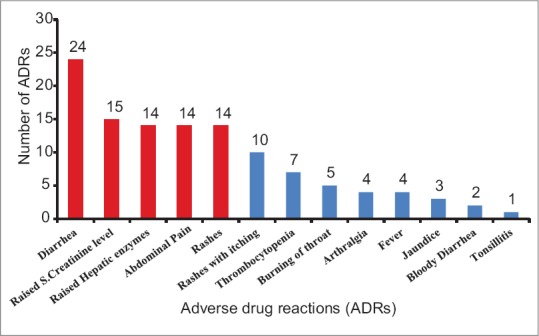
A total of 117 ADRs were observed during the study. These patients received a total of 19,498 doses (in 55 patients) of deferasirox during the study period. Most common ADRs observed were diarrhea (n = 24), raised serum creatinine (n = 15), raised hepatic enzymes (n = 14), abdominal pain (n = 14), and rashes (n = 14) [Figure 2]. No abnormalities were observed on ophthalmological examination at baseline and at end of the study.
| Figure 2: Adverse drug reactions observed with deferasirox in children with β thalassemia major at a tertiary care teaching hospital in India
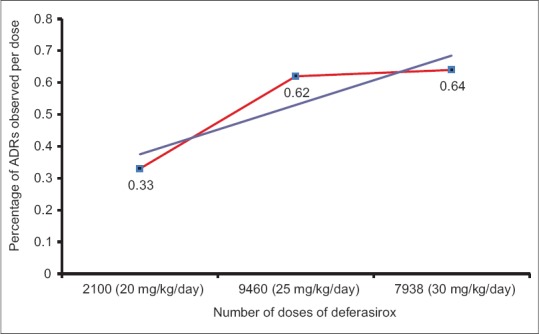
A total of seven ADRs were observed from 2100 doses of 20 mg/kg/day. Fifty-nine ADRs were reported from 9460 doses of 25 mg/kg/day and 51 ADRs from 7938 doses of 30 mg/kg/day of deferasirox. A positive correlation was observed between number of doses prescribed and percentage of ADRs observed (correlation coefficient [r] = 0.998, P = 0.03) [Figure 3].
| Figure 3: Correlation between number of doses of deferasirox and percent adverse drug reactions observed (r = 0.998, P = 0.03) in β thalassemic children reporting at a tertiary care teaching hospital in India (n = 55)
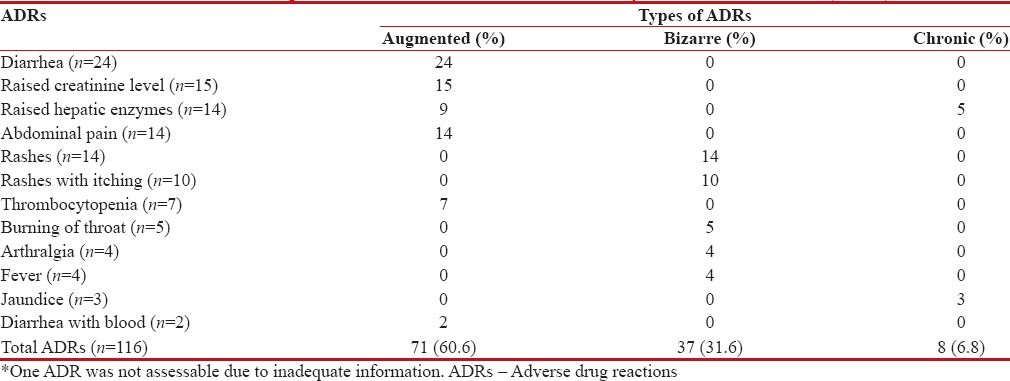
Types of ADRs observed were assessed based on available information on the safety of drug from clinical trials, postmarketing surveillance, and other studies. Majority of the ADRs observed were augmented (Type A) (n = 71, 60.6%), followed by bizarre (Type B) (n = 37, 31.6%) and chronic (Type C) (n = 8, 6.8%) [Table 5]. Tonsillitis occurring in one patient was not assessable due to the lack of adequate information.
Table 5
Adverse drug reactions observed with deferasirox in β thalassemic children (n=116)*
|
Hospitalization was required in 34 cases including 12 cases with raised serum creatinine level, ten cases with raised hepatic enzymes, seven cases of diarrhea, two cases of bloody diarrhea, and three cases of jaundice. These ADRs recovered with supportive treatment and temporary stoppage of the drug for a mean duration of 18 ± 8.2 days. Drug withdrawal was also indicated in seven cases of thrombocytopenia. The dose of deferasirox was reduced in 32 cases, to reduce the severity of ADRs. These ADRs also subsided following temporary modification of dose of deferasirox.
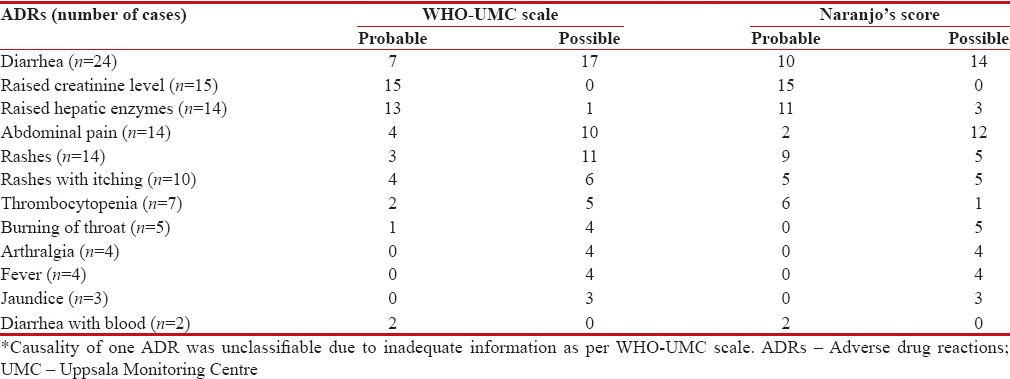
Deferasirox was the probable cause in 51 ADRs, while a possible cause in 65 ADRs as assessed by the WHO-UMC scale. Causality of deferasirox was unclassifiable in one case of tonsillitis. As per the Naranjo's score, deferasirox was the probable cause in sixty ADRs, while a possible causal relation with the drug was found in 57 ADRs including one case of tonsillitis [Table 6].
Table 6
Causality assessment of adverse drug reactions observed with deferasirox in β-thalassemic pediatric patients at Tertiary Care Teaching Hospital in India (n=116)*
|
Majority of ADRs (n = 83, 70.9%) were mild (severity level of 1 and 2) in nature, while 34 ADRs (29.1%) were of moderate severity (severity level of 3 and 4b) as assessed by the modified Hartwig and Siegel scale. Preventability of ADRs was assessed by modified Schumock and Thornton criteria. From a total of 117 ADRs, 110 (94.1%) ADRs were definitely preventable, while 7 (5.9%) ADRs were probably preventable.
Discussion
Thalassemia, a heterogeneous group of inherited disorders of hemoglobin synthesis, is the world's most common monogenic diseases. An estimated 80–90 million people in the world carry the beta thalassemia trait.[7] The disease is also common in India with approximately 10,000–12,000 children born every year with β thalassemia major.[8] India contributes to approximately 10% of the global disease burden.[9] Defective production of β globin chains in β thalassemia leads to an increased production of α globin chains. These globin chains get precipitated in RBCs, leading to extensive hemolysis and anemia. Resultant hypoxia and increased erythropoietin production cause expansion of ineffective erythroid mass. Definitive treatment of thalassemia includes bone marrow transplantation and gene therapy, both of which are expensive. In resource-limited settings, repeated BTs remain the mainstay of management. However, repeated BTs lead to iron overload and deposition of iron in various tissues of the body.[10] Iron overload is associated with a variety of complications affecting skeletal, cardiovascular, hepatobiliary, and endocrine systems. Hepatotoxicity due to iron overload is one of the leading causes of death in patients suffering from thalassemia.[11] To prevent these complications, iron chelation therapy is now recommended and routinely prescribed to transfusion-dependent thalassemic patients.[12]
Conventional iron chelators, i.e., desferioxamine and deferiprone, are effective in reducing iron overload. However, these drugs, owing to a shorter half-life, require frequent administration. Furthermore, desferioxamine administered by intravenous or subcutaneous route causes discomfort and affects patient compliance. Deferasirox is a newer iron chelator, effective orally. The drug requires once-daily administration. Efficacy and safety of deferasirox in children have been reported to be similar to that in adults.[13] However, data regarding efficacy and safety of this drug in Indian population are yet lacking. Hence, the present study was conducted to evaluate the utilization pattern, efficacy, safety, and tolerability of deferasirox in transfusion-dependent pediatric patients of β thalassemia.
During the study period, 10,980 pediatric patients were admitted annually at the study center. The prevalence of β thalassemia major in these patients was found to be about 4%. In the 55 patients enrolled, a male preponderance (65.45%) was observed. A similar preponderance of males (69.5%) was observed in studies conducted at Orissa, India[14] and at Faisalabad, Pakistan (65.66%).[15] The reason for male gender dominance in this disease has not been documented. However, in our study, this could be due to deep-rooted gender bias in favor of male child in India among parents. The parents are known to seek medical care more frequently and are willing to spend more for care of the male child.[16] Hence, whether this observation reflects a true male preponderance of the disease requires further evaluation.
Majority of patients in the present study were diagnosed with thalassemia major between the ages of 6 and 12 months, with a mean age at diagnosis of 14.05 ± 9.8 months. These findings are comparable to the observations made by Nigam et al.[17] who reported a mean age at diagnosis of thalassemia patients in Gujarat to be 12.7 months. Availability of advanced diagnostic techniques, media attention, increased awareness, and better access to healthcare has helped screen and diagnose this disease at an early age.
In the present study, a history of consanguineous marriage in parents was evident in nearly 65% patients. These patients belonged to Devipujak (23.6%), Muslim (16.3%), Sindhi (10.9%), and Chamar (10.9%) castes. Higher prevalence of thalassemia major in Muslim, Sindhi, Devipujak, Liana, and Chamar communities has been reported by other researchers too.[18] Practice of consanguineous marriages in these communities may be a major factor responsible for higher prevalence of thalassemia in these communities. An increased awareness of the consequences of consanguineous marriage and premarital and preconception screening for thalassemia trait should be encouraged to reduce the risk of thalassemia major in the offspring.
In the present study, a low LVEF was observed in about 16% patients at the time of enrolment. Patients of β thalassemia are prone to develop high-output cardiac failure due to prolonged tissue hypoxia secondary to chronic anemia, presence of hemoglobin F, and low levels of 2, 3-bisphosphoglycerate in the transfused blood.[19] Furthermore, repeated BTs in these patients lead to a large amount of free iron in plasma which can enter the cardiac myocytes, lead to peroxidative damage and myocyte apoptosis,[19] and worse the cardiac function. However, a significant improvement in the mean LVEF was observed in patients showing subnormal LVEF at the time of initiation of deferasirox. Here, further deterioration of cardiac function was probably prevented by effective iron chelation. Studies conducted by Wood[20] and Pennell et al.[21] have shown that LVEF of patients treated with deferasirox remained stable over a period of 2 years.
In the present study, each patient received an average of 1.5 units of PCV per month. Nonsplenectomized patients received an average 180.3 ml/kg/year of PCV and splenectomized patients received an average 131.8 ml/kg/year of PCV. This was in accordance with IAP guidelines,[4] which recommends that patient of β thalassemia major should receive 15–18 units of PCV, at the rate of 10–15 ml/kg annually. The volume to be transfused is 180 ml/kg/year of PCV in nonsplenectomized patients and 133 ml/kg/year in splenectomized patients.
In the present study, the mean serum ferritin level was detected to be significantly higher in patients of 7–10 and 11–12 years as compared to that in patients of 1–3 years at baseline. Mean serum iron level at baseline was also significantly higher in the higher age groups as compared to the age group of 1–3 years. Higher mean serum ferritin and serum iron levels in these patients could have been because of a higher number of BTs received in these patients before enrolment. With deferasirox treatment, a serial and significant decrease in mean serum ferritin and mean serum iron levels were observed in all age groups. Furthermore, a significant negative correlation was observed between the mean dose of deferasirox prescribed and the mean serum ferritin and mean serum iron levels. These findings suggest that deferasirox effectively reduced iron overload. A phase III study also showed a significant decrease in serum ferritin levels (mean reduction of 1137 ± 453 ng/ml) with deferasirox treatment (30 mg/kg/day) in 296 transfusion-dependent β thalassemia patients over a period of 1 year. In the same study, serum ferritin levels were stabilized at 20 mg/kg/day of deferasirox.[13] Pennell et al. also reported a median reduction of 1048 ng/ml in serum ferritin from baseline over 1 year in transfusion-dependent β thalassemic children after treatment with deferasirox at doses of 20–30 mg/kg/day.[22]
In the present study, all patients received deferasirox in doses ranging from 20 to 30 mg/kg/day. While adherence to drug administration guidelines[4] was observed in majority of the patients, concomitant use of food and improper dispersion of deferasirox was observed in 27.3% patients. This can be avoided by education of patients and caretakers regarding proper administration of deferasirox.
In the present study, the mean number of BTs received was higher in age groups of more than 3 years at baseline as compared to children aged 1–3 years. A significantly higher mean dose of deferasirox was prescribed to patients in higher age groups at baseline as compared to those aged 1–3 years. Furthermore, in each age group, the mean dose of deferasirox prescribed at the end of study period was significantly higher as compared to that at the baseline. The number of BT increases with age of the patient. This also increases iron overload proportionately. Hence, the requirement of a higher dose of deferasirox in these age groups was justified. An increment in dose of deferasirox was employed in patients of 1–3 years and 4–6 years to reduce iron overload and achieve target serum ferritin levels. High-serum ferritin levels require dose increment in older children; however, this was not observed in the present study.
ADRs were observed in nearly 95% patients. Higher incidence of ADRs in these patients can be attributed to the prolonged and continuous use of deferasirox. A positive correlation between the dose of deferasirox prescribed and the number of ADRs observed per dose was present. Taher et al. have also reported a higher incidence of ADRs in patients receiving 30 mg/kg/day deferasirox as compared to those receiving a daily dose of 20 mg/kg.[23] Most common ADR in the present study was diarrhea with or without blood (22.2%). Diarrhea was reported as the most common ADR with deferasirox in two phase II clinical trials and reported incidence of diarrhea in these studies (27% and 30%) was similar to that observed in the current study.[24] Diarrhea in these patients can be because of a faulty drug administration technique causing improper drug dispersion.[4] In the present study, a history of nonadherence to deferasirox administration guidelines was present in more than 50%-cases of diarrhea in the form of either inadequate dispersion of deferasirox tablet or concomitant use of food. Emphasis should therefore be laid on educating the patient/caretakers at each visit to ensure better drug compliance and to reduce the incidence of adverse reactions such as diarrhea.
Other common ADRs in the present study were rashes with or without itching (20.5%), raised serum creatinine level (12.8%), raised transminase levels (11.9%), and abdominal pain (11.9%). Raised serum creatinine (38%) and skin rash (10.8%) were the common ADRs reported with deferasirox by some authors.[13] A phase II trial has reported a higher incidence of abdominal pain (20%), raised transaminase level (20%), and tonsillitis (15%) with deferasirox.[2] However, the incidence of these ADRs was comparatively less in the present study which may be because of the smaller sample size. Thrombocytopenia (6%), burning of throat (4.2%), arthralgia (3.4%), fever (3.4%), jaundice (2.5%), and tonsillitis (0.8%) were the other ADRs observed in the present study.
In the present study, 60%-ADRs required a temporary withdrawal or reduction in the dose of deferasirox. These ADRs included diarrhea with or without blood, raised serum creatinine level, raised serum transaminase level, abdominal pain, and thrombocytopenia. Most of the observed ADRs were augmented (Type A) reactions. The suspect bizarre (Type B) reactions observed in the present study included rashes, burning throat, arthralgia, fever, and tonsillitis are unrelated to the pharmacological action of the deferasirox. Bizarre reactions reported in other studies include fever, arthralgia, nasopharyngitis, and rash. Pennell et al. reported that fever and arthralgia accounted for 3.5% and 2.9% of all ADRs with deferasirox, respectively.[22] Wood et al. reported that patients treated with this drug suffered from nasopharyngitis[25] and this accounts for 7.4% for all ADRs reported in these patients. Raised hepatic enzyme (serum transaminase) and jaundice, observed in nearly 31% patients in the present study, were chronic (Type C) reactions. Deferasirox is primarily metabolized through phase II conjugation in the liver. This may be responsible for hepatic injury due to this drug.[26]
Hospitalization was required in 29%-cases of ADRs in the present study, which included raised serum creatinine, raised transaminase, diarrhea with or without blood, and jaundice. These ADRs recovered with supportive treatment and temporary withdrawal of the drug for a mean duration of 18 ± 8.2 days. Cappellini et al. had also reported diarrhea, raised serum creatinine, and raised serum transaminase as the most common dose-dependent ADRs with deferasirox which require temporary withdrawal of drug.[13] Majority (94.1%) of ADRs observed were definitely preventable. These observations suggest that deferasirox was well tolerated and relatively safe in transfusion-dependent pediatric patients of thalassemia. Majority of the ADRs were reported with higher doses of deferasirox, suggesting the need for routine monitoring of serum ferritin and maintaining the patients on lowest effective dose possible. An awareness among prescribers about ADRs of deferasirox and frequent monitoring of serum ferritin and serum iron levels as a guide to determine its dose is required to improve tolerability of this important and necessary drug.
Conclusion
β thalassemia major is more common in males in this study. An improved awareness regarding consequences of consanguineous marriage, importance of prenatal screening of thalassemia trait, and genetic counseling of couples affected with thalassemia trait is needed. A significant decrease in the mean serum ferritin and serum iron levels in all age groups along with maintenance of cardiac function within normal limits is observed with deferasirox treatment. Deferasirox is relatively well tolerated among these patients. Deferasirox is least tolerated in the dose of 30 mg/kg/day. Periodic monitoring of laboratory and clinical parameters along with suitable dose modification can help optimize the drug therapy and improve the safety of this drug. Caution should be exercised for the right method of drug administration to improve efficacy and minimize ADRs due to deferasirox.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We would like to thank the patients and their parents/guardians for their consent and cooperation for the study.
- Benz EJ. Hemoglobinopathies. In: Longo D, Kasper D, Jameson J, Fauci A, Hauser S, Loscalzo J, editors. Harrison's Principles of Internal Medicine. 18th ed. New York: McGraw-Hill; 2012. p. 698-702.
- Galanello R, Piga A, Forni GL, Bertrand Y, Foschini ML, Bordone E, et al. Phase II clinical evaluation of deferasirox, a once-daily oral chelating agent, in pediatric patients with beta-thalassemia major. Haematologica 2006;91:1343-51.
- Maheshwari M, Arora S, Kabra M, Menon PS. Carrier screening and prenatal diagnosis of beta-thalassemia. Indian Pediatr 1999;36:1119-25.
- Indian Academy of Pediatrics. Guidelines for Diagnosis and Management of Thalassemia. IAP National Consensus Meeting on Thalassemia Held under IAP Action Plan; June, 2006. p. 169-202.
- Hoffbrand AV, Taher A, Cappellini MD. How I treat transfusional iron overload. Blood 2012;120:3657-69.
- Borgna-Pignatti C, Rugolotto S, De Stefano P, Piga A, Di Gregorio F, Gamberini MR, et al. Survival and disease complications in thalassemia major. Ann N Y Acad Sci 1998;850:227-31.
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis 2010;5:11.
- Gorakshakar AC, Colah RB. Cascade screening for beta-thalassemia: A practical approach for identifying and counseling carriers in India. Indian J Community Med 2009;34:354-6.
- Varawalla NY, Old JM, Sarkar R, Venkatesan R, Weatherall DJ. The spectrum of beta-thalassaemia mutations on the Indian subcontinent: The basis for prenatal diagnosis. Br J Haematol 1991;78:242-7.
- Olivieri NF, Brittenham GM. Iron-chelating therapy and the treatment of thalassemia. Blood 1997;89:739-61.
- ;Zurlo MG, De Stefano P, Borgna-Pignatti C, Di Palma A, Piga A, Melevendi C, et al. Survival and causes of death in thalassaemia major. Lancet 1989;2:27-30.
- Brittenham GM. Iron-chelating therapy for transfusional iron overload. N Engl J Med 2011;364:146-56.
- Cappellini MD, Cohen A, Piga A, Bejaoui M, Perrotta S, Agaoglu L, et al. Aphase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 2006;107:3455-62.
- Chhotray GP, Dash BP, Ranjit M. Spectrum of hemoglobinopathies in Orissa, India. Hemoglobin 2004;28:117-22.
- ;Ahmad L, Hassan M, Rana SM, Mahboob S, Jabeen F. Prevalence of β-thalassemic patients associated with consanguinity and anti-HCV antibody positivity – A cross sectional study. Pak J Zool 2011;43:29-36.
- Talsania S, Talsania N, Nayak H. A cross sectional study of thalassemia in Ahmedabad city, Gujarat. (Hospital based). Healthline 2011;2:48-51.
- Nigam N, Musnhi N, Patel M, Soni A. Distribution of β thalassemia and its correlation with α thalassemia in Gujarati families. Int J Hum Genet 2003;3:221-4.
- Shah N, Mishra A, Chauhan D, Vora C, Shah NR. Study on effectiveness of transfusion program in thalassemia major patients receiving multiple blood transfusions at a transfusion centre in Western India. Asian J Transfus Sci 2010;4:94-8.
- Tanner MA, Galanello R, Dessi C, Smith GC, Westwood MA, Agus A, et al. Arandomized, placebo-controlled, double-blind trial of the effect of combined therapy with deferoxamine and deferiprone on myocardial iron in thalassemia major using cardiovascular magnetic resonance. Circulation 2007;115:1876-84.
- Wood JC. Cardiac iron across different transfusion-dependent diseases. Blood Rev 2008;22 Suppl 2:S14-21.
- Pennell DJ, Porter JB, Cappellini MD, El-Beshlawy A, Chan LL, Aydinok Y, et al. Efficacy of deferasirox in reducing and preventing cardiac iron overload in beta-thalassemia. Blood 2010;115:2364-71.
- Pennell DJ, Porter JB, Cappellini MD, Chan LL, El-Beshlawy A, Aydinok Y, et al. Continued improvement in myocardial T2* over two years of deferasirox therapy in ß-thalassemia major patients with cardiac iron overload. Haematologica 2011;96:48-54.
- Taher A, El-Beshlawy A, Elalfy MS, Al Zir K, Daar S, Habr D, et al. Efficacy and safety of deferasirox, an oral iron chelator, in heavily iron-overloaded patients with beta-thalassaemia: The ESCALATOR study. Eur J Haematol 2009;82:458-65.
- Piga A, Galanello R, Forni GL, Cappellini MD, Origa R, Zappu A, et al. Randomized phase II trial of deferasirox (Exjade, ICL670), a once-daily, orally-administered iron chelator, in comparison to deferoxamine in thalassemia patients with transfusional iron overload. Haematologica 2006;91:873-80.
- Wood JC, Kang BP, Thompson A, Giardina P, Harmatz P, Glynos T, et al. The effect of deferasirox on cardiac iron in thalassemia major: Impact of total body iron stores. Blood 2010;116:537-43.
- Aslam N, Mettu P, Marsano-Obando LS, Martin A. Deferasirox induced liver injury in haemochromatosis. J Coll Physicians Surg Pak 2010;20:551-3.
References
| Figure 1Age and gender distribution of children with β thalassemia major reporting at a tertiary care teaching hospital (n = 55)
| Figure 2: Adverse drug reactions observed with deferasirox in children with β thalassemia major at a tertiary care teaching hospital in India
| Figure 3: Correlation between number of doses of deferasirox and percent adverse drug reactions observed (r = 0.998, P = 0.03) in β thalassemic children reporting at a tertiary care teaching hospital in India (n = 55)
- Benz EJ. Hemoglobinopathies. In: Longo D, Kasper D, Jameson J, Fauci A, Hauser S, Loscalzo J, editors. Harrison's Principles of Internal Medicine. 18th ed. New York: McGraw-Hill; 2012. p. 698-702.
- Galanello R, Piga A, Forni GL, Bertrand Y, Foschini ML, Bordone E, et al. Phase II clinical evaluation of deferasirox, a once-daily oral chelating agent, in pediatric patients with beta-thalassemia major. Haematologica 2006;91:1343-51.
- Maheshwari M, Arora S, Kabra M, Menon PS. Carrier screening and prenatal diagnosis of beta-thalassemia. Indian Pediatr 1999;36:1119-25.
- Indian Academy of Pediatrics. Guidelines for Diagnosis and Management of Thalassemia. IAP National Consensus Meeting on Thalassemia Held under IAP Action Plan; June, 2006. p. 169-202.
- Hoffbrand AV, Taher A, Cappellini MD. How I treat transfusional iron overload. Blood 2012;120:3657-69.
- Borgna-Pignatti C, Rugolotto S, De Stefano P, Piga A, Di Gregorio F, Gamberini MR, et al. Survival and disease complications in thalassemia major. Ann N Y Acad Sci 1998;850:227-31.
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis 2010;5:11.
- Gorakshakar AC, Colah RB. Cascade screening for beta-thalassemia: A practical approach for identifying and counseling carriers in India. Indian J Community Med 2009;34:354-6.
- Varawalla NY, Old JM, Sarkar R, Venkatesan R, Weatherall DJ. The spectrum of beta-thalassaemia mutations on the Indian subcontinent: The basis for prenatal diagnosis. Br J Haematol 1991;78:242-7.
- Olivieri NF, Brittenham GM. Iron-chelating therapy and the treatment of thalassemia. Blood 1997;89:739-61.
- ;Zurlo MG, De Stefano P, Borgna-Pignatti C, Di Palma A, Piga A, Melevendi C, et al. Survival and causes of death in thalassaemia major. Lancet 1989;2:27-30.
- Brittenham GM. Iron-chelating therapy for transfusional iron overload. N Engl J Med 2011;364:146-56.
- Cappellini MD, Cohen A, Piga A, Bejaoui M, Perrotta S, Agaoglu L, et al. Aphase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 2006;107:3455-62.
- Chhotray GP, Dash BP, Ranjit M. Spectrum of hemoglobinopathies in Orissa, India. Hemoglobin 2004;28:117-22.
- ;Ahmad L, Hassan M, Rana SM, Mahboob S, Jabeen F. Prevalence of β-thalassemic patients associated with consanguinity and anti-HCV antibody positivity – A cross sectional study. Pak J Zool 2011;43:29-36.
- Talsania S, Talsania N, Nayak H. A cross sectional study of thalassemia in Ahmedabad city, Gujarat. (Hospital based). Healthline 2011;2:48-51.
- Nigam N, Musnhi N, Patel M, Soni A. Distribution of β thalassemia and its correlation with α thalassemia in Gujarati families. Int J Hum Genet 2003;3:221-4.
- Shah N, Mishra A, Chauhan D, Vora C, Shah NR. Study on effectiveness of transfusion program in thalassemia major patients receiving multiple blood transfusions at a transfusion centre in Western India. Asian J Transfus Sci 2010;4:94-8.
- Tanner MA, Galanello R, Dessi C, Smith GC, Westwood MA, Agus A, et al. Arandomized, placebo-controlled, double-blind trial of the effect of combined therapy with deferoxamine and deferiprone on myocardial iron in thalassemia major using cardiovascular magnetic resonance. Circulation 2007;115:1876-84.
- Wood JC. Cardiac iron across different transfusion-dependent diseases. Blood Rev 2008;22 Suppl 2:S14-21.
- Pennell DJ, Porter JB, Cappellini MD, El-Beshlawy A, Chan LL, Aydinok Y, et al. Efficacy of deferasirox in reducing and preventing cardiac iron overload in beta-thalassemia. Blood 2010;115:2364-71.
- Pennell DJ, Porter JB, Cappellini MD, Chan LL, El-Beshlawy A, Aydinok Y, et al. Continued improvement in myocardial T2* over two years of deferasirox therapy in ß-thalassemia major patients with cardiac iron overload. Haematologica 2011;96:48-54.
- Taher A, El-Beshlawy A, Elalfy MS, Al Zir K, Daar S, Habr D, et al. Efficacy and safety of deferasirox, an oral iron chelator, in heavily iron-overloaded patients with beta-thalassaemia: The ESCALATOR study. Eur J Haematol 2009;82:458-65.
- Piga A, Galanello R, Forni GL, Cappellini MD, Origa R, Zappu A, et al. Randomized phase II trial of deferasirox (Exjade, ICL670), a once-daily, orally-administered iron chelator, in comparison to deferoxamine in thalassemia patients with transfusional iron overload. Haematologica 2006;91:873-80.
- Wood JC, Kang BP, Thompson A, Giardina P, Harmatz P, Glynos T, et al. The effect of deferasirox on cardiac iron in thalassemia major: Impact of total body iron stores. Blood 2010;116:537-43.
- Aslam N, Mettu P, Marsano-Obando LS, Martin A. Deferasirox induced liver injury in haemochromatosis. J Coll Physicians Surg Pak 2010;20:551-3.