 PDF
PDF  Views
Views  Share
Share


Imaging Recommendations for Diagnosis, Staging, and Management of Hereditary Malignancies
CC BY 4.0 · Indian J Med Paediatr Oncol 2023; 44(03): 287-301
DOI: DOI: 10.1055/s-0042-1760325
Abstract
Hereditary cancer syndromes, characterized by genetically distinct neoplasms developing in specific organs in more than one family members, predispose an individual to early onset of distinct site-specific tumors. Early age of onset, multiorgan involvement, multiple and bilateral tumors, advanced disease at presentation, and aggressive tumor histology are few characteristic features of hereditary cancer syndromes. A multidisciplinary approach to hereditary cancers has led to a paradigm shift in the field of preventive oncology and precision medicine. Imaging plays a pivotal role in the screening, testing, and follow-up of individuals and their first- and second-degree relatives with hereditary cancers. In fact, a radiologist is often the first to apprise the clinician about the possibility of an underlying hereditary cancer syndrome based on pathognomonic imaging findings. This article focuses on the imaging spectrum of few common hereditary cancer syndromes with specific mention of the imaging features of associated common and uncommon tumors in each syndrome. The screening and surveillance recommendations for each condition with specific management approaches, in contrast to sporadic cases, have also been described.
Publication History
Article published online:
01 March 2023
© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Hereditary cancer syndromes, characterized by genetically distinct neoplasms developing in specific organs in more than one family members, predispose an individual to early onset of distinct site-specific tumors. Early age of onset, multiorgan involvement, multiple and bilateral tumors, advanced disease at presentation, and aggressive tumor histology are few characteristic features of hereditary cancer syndromes. A multidisciplinary approach to hereditary cancers has led to a paradigm shift in the field of preventive oncology and precision medicine. Imaging plays a pivotal role in the screening, testing, and follow-up of individuals and their first- and second-degree relatives with hereditary cancers. In fact, a radiologist is often the first to apprise the clinician about the possibility of an underlying hereditary cancer syndrome based on pathognomonic imaging findings. This article focuses on the imaging spectrum of few common hereditary cancer syndromes with specific mention of the imaging features of associated common and uncommon tumors in each syndrome. The screening and surveillance recommendations for each condition with specific management approaches, in contrast to sporadic cases, have also been described.
Introduction
Hereditary cancer syndromes (HCSs) are syndromes characterized by genetically distinct neoplasms developing in specific organs in more than one family members,[1] [2] predisposing an individual to early onset of distinct site-specific tumors. A radiologist is often the first to apprise the clinician about the possibility of an underlying HCSs based on pathognomonic imaging findings, leading to genetic testing of the individual and their relatives as a part of preventive oncology. Radiogenomics is a term coined in recent years that describes the relationship between imaging features of a lesion and the underlying genetic or molecular abnormality, which aides in approach to diagnosis, guiding therapeutic strategies, assessment of treatment response, and prognostication.[3]
Most of the HCSs have an autosomal dominant pattern of inheritance and comprise up to 5 to 10%-of the overall worldwide cancer burden.[1] [2] Among the Indian population, there is paucity of data on the incidence of hereditary cancers. The mortality rates are possibly higher in India, attributed to different lifestyles, delayed introduction of screening programs, and socioeconomic limitation to treatment access.[4] The lack of knowledge and access to accurate information as well as the social taboo and stigma associated with cancers in India leads to under-reporting and contributes to the skewed incidence.[5]
The different genetic mechanisms that play a role in the etiopathogenesis of HCSs include inactivation of tumor suppressor genes or reactivation of proto-oncogenes or abnormalities in the DNA repair genes.[6] Predisposition to certain malignancies and the therapeutic response are determined by the interaction of these genetic mutations with environmental factors called gene–environment interaction.[7] Early age of onset, multiorgan involvement, multiple and bilateral tumors, advanced disease at presentation, and aggressive tumor histology are characteristic of HCSs. A fundamental change in the approach to treatment of hereditary cancers and a paradigm shift to preventive oncology is proof that a multidisciplinary approach with collaboration of clinical, radiological, and pathological specialties with genetic research is a promising step toward precision medicine. The common and uncommon tumors observed in few commonly encountered HCS and their associated mutations have been summarized in [Table 1]. Several childhood cancers are also associated with germline and somatic mutations in cancer predisposition genes. The Society for Pediatric Oncology and Hematology established a CPS (cancer predisposition syndrome) working group that summarized a comprehensive review of childhood CPS and provided clinical and diagnostic recommendations for cancer prevention, surveillance, treatment, and follow-up.[8] Few of the common ones are summarized in [Table 2].
|
Predisposition group |
Genes involved |
Associated cancers |
|---|---|---|
|
Ataxia telangiectasia |
ATM |
Leukemia |
|
Lymphoma |
||
|
Bloom syndrome |
BLM |
Leukemia |
|
Lymphoma |
||
|
Constitutional mismatch repair-deficiency syndrome (CMMR-D) |
MLH1, MSH2, MSH6, PMS2 |
Pediatric brain tumors |
|
Colorectal cancers |
||
|
ALL, AML, lymphoma |
||
|
Early onset GI and GU tumors |
||
|
Fanconi anemia |
FANCA, C, D1, D2, E, F, G, I, J, L, M, RAD51C, SLX4/BTBD12, FANCAB |
Leukemia (MDS, AML) |
|
Squamous cell carcinoma |
||
|
Wilms tumor |
||
|
Gynecological tumors |
||
|
Brain tumors |
||
|
Wiskott-Aldrich syndrome (WAS) |
WAS |
Diffuse large B cell lymphoma |
|
Non-Hodgkin's lymphoma of larynx |
||
|
Cerebellar astrocytoma |
||
|
Kaposi sarcoma |
||
|
Smooth muscle tumors |
||
|
Wilms-Aniridia-GU-anomaly-retardation (WAGR) syndrome |
WT1 |
Wilms tumor |
|
Gonadoblastoma |
||
|
Denys-Drash syndrome (DDS) |
WT1 (dominant) |
Wilms tumor |
|
Gonadoblastoma |
||
|
Beckwith-Wiedemann syndrome (BWS) |
p57, H19, LIT1, ICR1, CDKN1C, NSD1 |
Wilms tumor |
|
Hepatoblastoma |
||
|
Adrenal carcinoma |
||
|
Rhabdomyosarcoma |
||
|
Familial pleuropulmonary blastoma tumor predisposition syndrome |
DICER1 |
Pleuropulmonary blastoma |
|
Cystic nephroma |
||
|
Sertoli-Leydig cell tumors |
||
|
Rhabdomyosarcoma |
||
|
Supratentorial primitive neuroectodermal tumor |
||
|
Intraocular medulloepithelioma |
||
|
Nevoid basal cell carcinoma syndrome (NBCCS)/Gorlin syndrome |
PTCH1, 2, SUFU |
Basal cell carcinoma |
|
Desmoplastic medulloblastoma |
||
|
Ovarian fibromas |
||
|
Familial retinoblastoma syndrome (RB) |
RB1 |
Retinoblastoma |
|
Osteosarcoma |
||
|
Melanoma |
||
|
Glioma |
||
|
Rhabdoid tumor predisposition syndrome |
SMARCB1/INI1 |
Rhabdoid tumor |
|
Medulloblastoma |
||
|
Choroid plexus tumor |
||
|
Schwannoma |
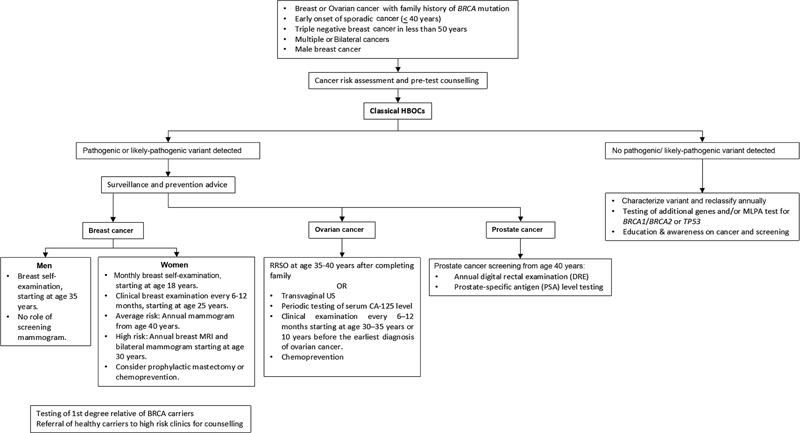
| Fig 1 :Imaging and clinical screening recommendations in BRCA carriers. CT, computed tomography; HBOCS, hereditary breast and ovarian cancer syndrome; MRI, magnetic resonance imaging; RRSO, risk-reducing salpingo-oophorectomy.
Li-Fraumeni Syndrome
Two criteria exist for diagnosis of Li-Fraumeni syndrome (LFS), the classic and Chompret criteria, summarized in [Fig. 2], the latter having a higher sensitivity (82–95%) and specificity (47–58%).[27] The lifetime risk of developing cancer with TP53 mutation is nearly 100%-for females and 73%-for males.[28] The incidence of tumors in LFS is 27 and 16% for soft tissue sarcomas (most common: rhabdomyosarcoma) and osteosarcomas, 60%-for breast cancer, 13%-for brain and adrenocortical cancers, and 4%-for leukemias, respectively.[27] [29] Although radiation-induced cancers are rare in the normal population (accounting for less than 5%-of all treatment-related cancers), individuals with LFS have increased susceptibility to DNA-damaging effects of ionizing radiation.[30] They are at high risk of developing secondary malignancies in a previously radiated field, with an incidence of 30%-and a median time for the development of 10.7 years.[27] In cancer patients testing for TP53 variants, surgical and ablative therapeutic measures are the preferred first-line treatment while advocating the use of non-genotoxic chemotherapeutic agents and avoiding radiotherapy when possible.[31] However, a recent study done by Hendrickson et al did find that the rate of a subsequent malignancy was not significantly different between LFS patients who received radiation compared with those who did not. In fact, none of the subsequent malignancies in patients receiving radiation could be confidently classified as radiation-associated malignancy and were merely considered as disease recurrence.[32]
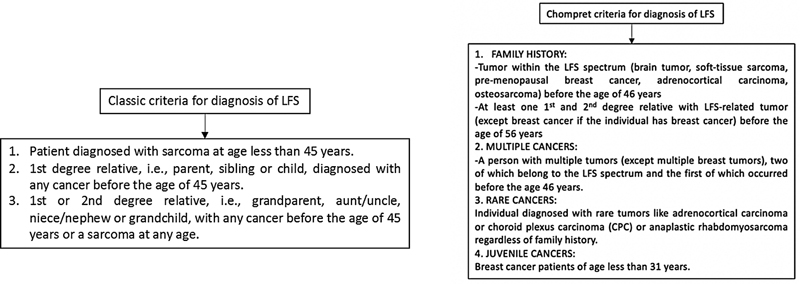
| Figure 2:Classic and Chompret criteria for diagnosis of Li-Fraumeni syndrome (LFS).
Previously, studies have evaluated the use of 2-deoxy-2-[fluorine-18]fluoro- D-glucose integrated with computed tomography (18F-FDG-PET/CT) as a screening tool for LFS; however, increased susceptibility to adverse effects of ionizing radiation negates routine use.[33] Whole body MRI (WB-MRI) now plays a pivotal role in surveillance of high-risk individuals. A meta-analysis done by Ballinger et al in 2017 estimated the cancer detection rate of WB-MRI as 7%-with a false-positive rate of 43%, stating that WB-MRI offers clinical utility in the baseline clinical risk management.[34] A study done by Villani et al proved that screening with WB-MRI, in adults and children with LFS, detected all tumors in asymptomatic individuals with a 3-year survival rate of 100%, as opposed to 23%-in persons who did not undergo surveillance.[35] The cancer detection rate in children with WB-MRI, however, may be lower than adults.[36] Image sequences include coronal T1 and short tau inversion recovery with axial diffusion-weighted images (DWI). Contrast administration is not routinely recommended.
Screening for breast cancers in LFS is done with annual breast MRI. Diagnostic imaging may include mammography but is reserved for older women and is not recommended for young women as the sensitivity of mammography is significantly lower in dense breasts of young women. Breast cancers are usually intraductal type and appear as irregular spiculated solid masses on imaging. Malignant phyllodes is also frequently associated with LFS appearing as a large heterogeneous mass with cystic and necrotic areas of degeneration and marked peripheral and internal vascularity on ultrasound. Women with LFS who develop breast cancer are advised to have bilateral mastectomy rather than lumpectomy to reduce the risk of developing a second primary breast cancer. Also, administration of radiotherapy post lumpectomy is also a challenge since it carries a risk of developing cancer in the radiation field. There is no known elevated risk of male breast cancer.
Almost 50%-patients with choroid plexus carcinoma are associated with TP53 alteration.[37] Gliomas like astrocytoma, oligodendroglioma and glioblastoma multiforme are other common brain tumors occurring in LFS. All these have typical features on imaging similar to their sporadic counterparts. Atypical presentation like leptomeningeal spread or involvement of the posterior fossa are encountered.[38]
Pediatric patients with adrenocortical cancer almost always have TP53 mutations, appearing as large heterogeneously enhancing masses with metastasis to lungs, liver and lymph nodes on cross-sectional imaging. Screening abdominal ultrasounds are usually sufficient; however, MRI is more sensitive for smaller lesions.
Surveillance recommendations in carriers of germline disease-causing TP53 variants is depicted in [Fig. 3].[39]
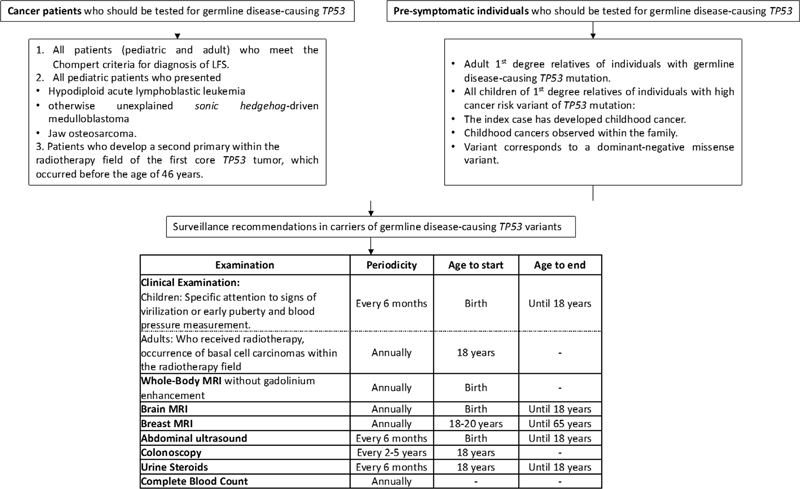
| Figure 3:Surveillance recommendations in carriers of germline disease-causing TP53 variants. LFS, Li-Fraumeni syndrome; MRI, magnetic resonance imaging.
von Hippel-Lindau Disease
von Hippel-Lindau Disease (VHL) is characterized by the development of vascular tumors due to inactivation of VHL gene that leads to upregulation of somatic and vascular growth factors, including vascular endothelial growth factor (VEGF). Renal, central nervous system, and pancreas are the most commonly involved organ systems.
The most common malignancy associated with VHL is renal cell carcinoma (RCC). Early diagnosis of RCC is imperative as they are known to be multiple, prone to relapse, and also the most common cause of death. Diagnosis as well as screening for RCCs utilizes the renal imaging protocol on CT (with unenhanced, cortico-medullary, nephrogenic, and excretory phases) which is the gold standard for depiction of characteristic vascular lesions showing arterial enhancement and venous wash out. MRI is increasingly preferred over CT for imaging in VHL owing to the absence of ionizing radiation, superior evaluation of smaller lesions, improved detection of intratumoral lipid, and hemorrhage and safer use with renal impairment. A recent study by Farhadi et al in 2020 correlated the relationship between apparent diffusion coefficient (ADC) and the growth rates of RCC for predicting the volume doubling time (VDT); ADC bearing a negative relationship with growth rate and positive relationship with VDT, thus, establishing the use of DWI in identifying RCCs with higher growth rates.[40] Imaging also plays an important role in targeted intervention. Percutaneous focal ablation, like CT-guided radio frequency ablation (RFA) and cryoablation, is the preferred treatment for RCCs in VHL as it significantly improves the cancer-free survival,[41] especially for smaller (< 3 href="https://www.thieme-connect.com/products/ejournals/html/10.1055/s-0042-1760325#JR22790732-41" xss=removed>41] Some prefer to use a cutoff of 4 cm for surgery as this may help delay the surgery time.[42]
About 70%-cases of VHL exhibit hemangioblastomas in the central nervous system, usually multiple with a high rate of recurrence, seen with decreasing frequency in the cerebellum (52%), spinal cord (cervical or thoracic) (44%), and brain stem (18%).[43] On MRI, intense hypervascular enhancement of mural nodule is seen with tumor-related cysts showing nonenhancing walls. Calcification is characteristically absent. Spinal MRI screening for spinal hemangioblastomas is mandatory in the presence of cerebellar hemangioblastoma as they usually coexist. Retinal hemangioblastomas are found in 70%-of cases over the age of 60 years. Diagnostic workup includes ophthalmoscopy and fluorescein angiography, the role of imaging being limited. MRI may depict a hypervascular retinal nodule with or without retinal detachment.
There are two subtypes of VHL, depending on the absence (type 1) and presence (type 2) of pheochromocytoma. VHL exhibits a distinct pattern of plasma catecholamine excess, almost exclusively norepinephrine production, as compared with combination of metanephrine and normetanephrine in MEN2 or NF1.[44] On cross-sectional imaging, pheochromocytomas show avid early arterial enhancement with venous washout. Calcification as well as areas of necrosis and hemorrhage may be seen. Radionuclide meta-iodobenzylguanidine (MIBG) scan can be used for tumor localization as well as detection of occult metastasis. Recent studies have showed that the diagnostic accuracy of WB-MRI is comparable to that of MIBG.[45]
Endolymphatic sac tumors are associated with VHL in mere 20%-cases; however, bilateral occurrence is pathognomic and can be detected on contrast-enhanced MRI of the brain and internal auditory meatus.
Pancreatic serous cystadenomas and neuroendocrine tumors (NET) are also common, with the latter harboring a malignant potential and detected in up to 15%- patients with VHL.[46] Serous cystadenomas have the typical appearance of “cluster of small cysts” with a central scar identified on cross-sectional imaging. They require no treatment unless symptomatic. NETs have the typical early arterial enhancement and venous washout like other hypervascular NET on cross-sectional imaging and are seen most commonly in the head and uncinate process of pancreas. Endoscopic ultrasound with or without contrast has a significant role in the diagnosis, intervention (tissue sampling and/or targeted ablation) and surveillance of NETs in VHL.[47] MRI has increased sensitivity over CT and the use of DWI has significantly improved the sensitivity of tumor detection, comparable to PET/CT wherein low ADC values suggest higher grade tumor.[47] 68Ga-DOTATATE PET/CT is useful for the depiction of small NETs, whereas 18F-FDG-PET/CT has increased sensitivity for higher grade tumors.[47]
Tumor-to-tumor metastasis is a peculiar phenomenon associated with VHL and refers to the spread of malignant cells from RCC, NET, and pheochromocytoma to existent hemangioblastomas, which act as a host for metastases,[48] suspected with sudden change in size of hemangioblastomas.
Systemic chemotherapeutic treatment in few VHL patients with inoperable or previously treated lesions is recommended, like VEGF-receptor inhibitor and fibroblast growth factor inhibitor.
Summary of individuals who should be tested for VHL mutation is given in [Fig. 4].

| Figure 4:Individuals who should be tested for VHL mutation. CNS, central nervous system; Hb, hemoglobin; PNET,—; RCC, renal cell carcinoma; VHL, von Hippel-Lindau disease.
VHL alliance suggested surveillance guidelines have been tabulated in [Fig. 5].[49] [50] [51]
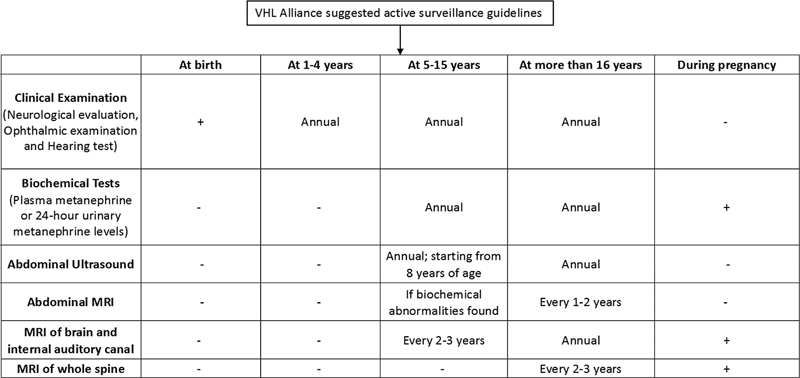
| Figure 5:VHL alliance suggested surveillance guidelines. MRI, magnetic resonance imaging; VHL, von Hippel-Lindau disease.
Multiple Endocrine Neoplasia Syndrome
Multiple endocrine neoplasia (MEN) syndrome is a group of autosomal dominant cancer syndromes characterized by the simultaneous presence of neoplasms in two or more endocrine organs.[52] There are two types: MEN 1 (Wermer's syndrome) and MEN2. MEN1 affects all age groups without any gender predilection with a prevalence of 2/100,000.[53] MEN2 has a prevalence of 1/200,000 live births.[54] The associated abnormalities in MEN syndromes have been summarized in [Fig. 6].
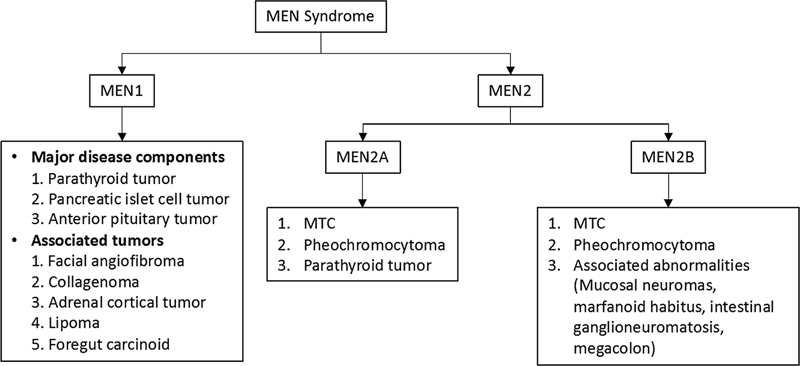
| Figure 6:Associated abnormalities with MEN syndromes. MEN, multiple endocrine neoplasia syndrome; MTC, medullary thyroid cancer.
MEN1
MEN1 constitutes many endocrine and nonendocrine tumors; however, approximately one-third of deaths in MEN1 are caused by endocrine malignancies. The crux of the management lies in early diagnosis and intervention that lead to improved outcome and decreased morbidity. Recommendations suggest performing genetic testing in individuals with significant family history or when the clinical presentation is suggestive of MEN1, supplemented with regular biochemical screening and cross-sectional imaging in patients who harbor the mutation.[55]
Parathyroid adenomas are the most common tumors in MEN1 (100%-cases), presenting with elevated parathormone and hypercalcemia, usually before the age of 20 to 25 years. On ultrasound, which is usually the first investigation, they appear as nodular hypoechoic masses with increased vascularity. Asymmetric uptake and retention on delayed images of 99mTc-sestamibi nuclear scintigraphy are pathognomic. The sensitivity and positive predictive values of ultrasound and nuclear scintigraphy are similar. Increased sensitivity of four-dimensional CT is observed especially with smaller lesions and multigland involvement.[56] Dynamic MRI is superior to CT for demonstrating the hypervascular nature of parathyroid adenomas. Contrast–time curve generation allows quantitative analysis of the perfusion parameters of parathyroid adenomas and reveals a faster time-to-peak, higher peak enhancement, and faster wash in washout compared with cervical lymph nodes or thyroid, thus improving the diagnostic accuracy.[57] Hyperparathyroidism is treated with surgery, usually entailing excision of three and a half gland (subtotal parathyroidectomy) in cases where all the four glands are affected.
Enteropancreatic NET, seen in 30 to 70%-cases, is a cause of significant morbidity and mortality in MEN1 syndrome as most of them are functional, however, with a lesser malignant potential. Transabdominal ultrasound is usually the first investigation performed. Endoscopic ultrasound has a higher sensitivity (100%) and specificity (95%), but is invasive and depends on operator efficiency. Cross-sectional imaging is most widely performed for diagnosis and staging. Most common are gastrinomas, frequently occurring in the duodenum, and tend to be multiple. Patients are usually offered non-surgical treatment except for cases with secretory tumors, pancreatic origin and size more than 2 cm. Patients with insulinoma, VIPoma, and glucagonoma are offered surgery.[58]
A high incidence of anterior pituitary tumors is observed in women with MEN1 (30–40%).[59] They are usually microadenomas (two-third cases) and best visualized on MRI, as small hypointense lesions on T1. Dynamic post-contrast sequence acquisition is essential for their depiction. 68Ga-DOTATATE PET/CT can identify functioning and nonfunctioning pituitary adenomas, although it is most useful in detecting residual normal pituitary tissue after adenomectomy and for diagnosis of recurrence of functioning adenomas.[60] Treatment remains similar to sporadic cases.
Screening protocol for MEN1 is depicted in [Fig. 7].[61]
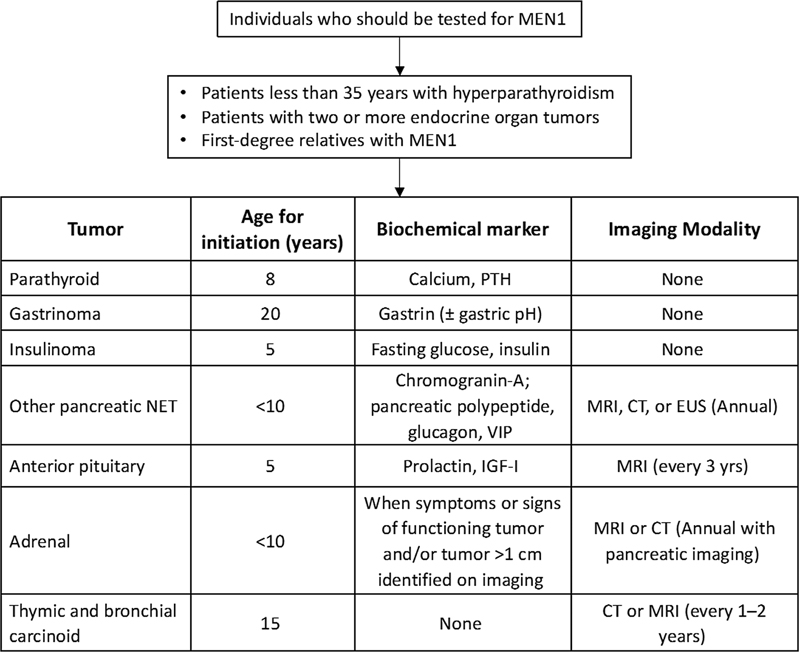
| Fig 7:Guidelines for screening protocol for MEN1 syndrome. CT, computed tomography; EUS, endoscopic ultrasound; IGF-1, insulin-like growth factor-1; MRI, magnetic resonance imaging; MEN1, multiple endocrine neoplasia 1; NET, neuroendocrine tumor; PTH, parathormone; VIP, vasoactive intestinal peptide.
MEN2
MEN2A (Sipple's syndrome) is more common and MEN2B is less common but more aggressive. All the variants of MEN2 show high penetrance of medullary thyroid cancer (MTC), seen in 90 to 95%-cases. Ultrasound should be performed for all patients with MTC as the initial evaluation. MTC appears as a solid markedly hypoechoic nodule with increased vascularity. Microcalcification may be seen. Metastasis to central and lateral compartment cervical nodes and mediastinal nodes with pulmonary and hepatic metastasis should be looked for. American Thyroid Association 2015 recommends contrast-enhanced CT scan of the neck and chest, triple-phase contrast-enhanced CT of the liver, axial MRI, and bone scintigraphy in cases with extensive neck disease or high calcitonin (>500 pg/dL) to rule out distant metastasis[55] in MTC cases. Recently, European Association of Nuclear Medicine guidelines 2020 recommended use of 18F-FDOPA PET/CT (6–18F-fluoro-L-3,4-dihydroxyphenylalanine) in persistent MTC. Use of 18F-FDG-PET/CT is limited to aggressive variants, while 68Ga-somatostatin analogs PET/CT is suboptimal.[62] Surgery is the best form of prevention as well as cure for MTC with regular postoperative monitoring of serum calcitonin to detect recurrence. Early thyroidectomy may lower the mortality from hereditary MTC to less than 5%-[63] with a more aggressive neck dissection for successful cure.
There may be an association of benign or malignant pheochromocytomas in MEN2 syndrome. Therefore, once the germline RET mutation is confirmed, it is important to rule out the presence of a pheochromocytoma. If cross-sectional imaging is nondiagnostic or depicts a unilateral disease, functional imaging like MIBG is suggested that has a higher sensitivity (87–90%). The screening for pheochromocytomas should be started in childhood, age of onset varying according to the risk category of mutation. Screening consists of measuring free plasma and 24-hour urine metanephrine and nor-metanephrine supplemented with cross sectional imaging if these levels are high. In cases with synchronous presentation of MTC and pheochromocytoma, pheochromocytoma should be treated first, followed by MTC.[64] Adrenalectomy is the treatment of choice following adrenal blockade.
Parathyroid tumors are seen in MEN2A and require investigations similar to MEN1 syndrome. Hyperparathyroidism does not develop in MEN2B, and thus screening is not required.
Guidelines for biochemical and radiological screening in individuals at high risk of developing MEN2 are depicted in [Fig. 8].[61]
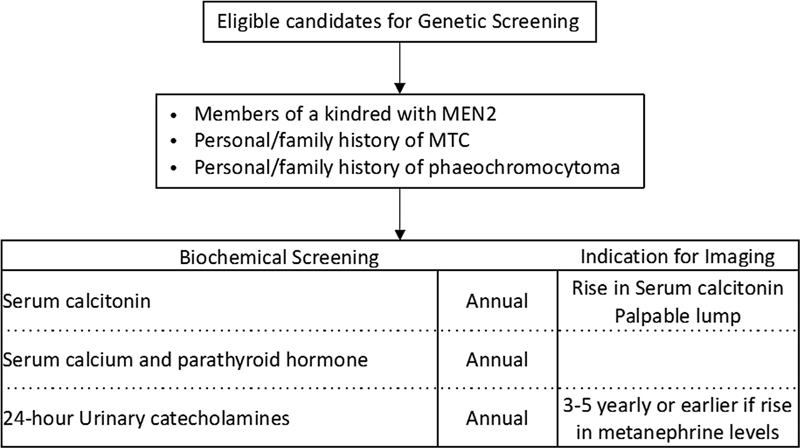
| fig 8:Guidelines for screening protocol for MEN2 syndrome. MEN2, multiple endocrine neoplasia 2; MTC, medullary thyroid cancer.
Tuberous Sclerosis
Tuberous sclerosis (TSC) is a neurocutaneous syndrome. The classic clinical triad for diagnosis includes facial adenoma sebaceum, epilepsy, and mental retardation, seen in 30 to 40%-cases. The clinical diagnosis of TSC is divided into major and minor features summarized in [Fig. 9].
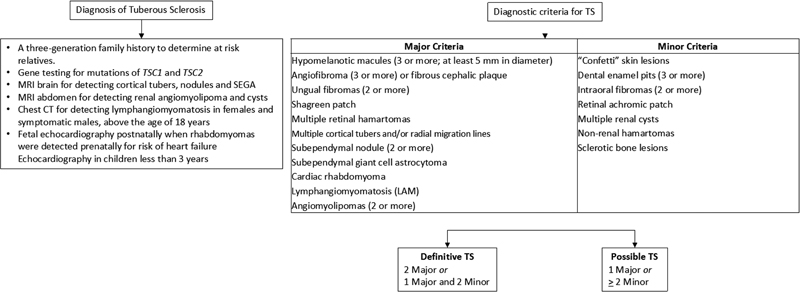
| fig 9:Clinical features for diagnosis of TSC. CT, computed tomography; SEGA, subependymal giant cell astrocytoma; TSC, tuberous sclerosis.
The most common intracranial manifestation of TSC is cortical tubers or hamartomas, seen in 95%-cases. On CT, they are hypodense areas of gyral widening. Calcification may be seen with progression. On MRI, they are T1 hypointense and T2 hyperintense with contrast enhancement in 10%-cases. Microstructural changes in the tubers are detected by diffusion tensor imaging.[65] Occasionally, a cyst-like appearance may be seen on MRI following evolution of cortical tubers. These are associated with more severe epilepsy phenotypes. Cortical tubers associated with refractory epilepsy often require surgery. Pulsed arterial spin-labeled technique helps in quantifying perfusion characteristics of cortical tubers. Hyperperfused lesions are associated with increased frequency of seizures.[66] Preoperative evaluation with 18F-FDG-PET/MRI for TSC has emerged as an indispensable noninvasive method to select the tubers as surgical candidates in refractory epilepsy.[67] Evaluating the involvement of perituberal cortex on MRI is essential for defining the extent of resection.[68]
Subependymal nodules are also common and appear isointense on T1 and iso- to hyperintense on T2 more and are more prone to calcification than cortical tubers. Radial migration lines are the most common white matter abnormality identified in TSC.
Subependymal giant cell astrocytoma (SEGA) is defined as a lesion more than 1 cm in size at the caudothalamic groove or a subependymal lesion showing serial growth.[69] It is the most common central nervous system tumor in TSC, seen in 10 to 15%-cases. It presents in late childhood and remains asymptomatic unless complicated with hydrocephalus. On MRI, they are hypo- to isointense compared with cortex on T1 and heterogeneously iso- to hyperintense on T2, most of them showing contrast enhancement. Early surgical resection is the standard treatment. Medical management with mechanistic target of rapamycin (mTOR) inhibitors is currently indicated for patients with symptomatic SEGA that are not amendable to surgery.[70]
Lymphangioleiomyomatosis (LAM) is characterized by multiple thin-walled cysts scattered in lungs. Strong female preponderance is noted with symptoms developing in the middle age. TSC association is seen in approximately 15%-cases of LAM. Solitary or multiple cardiac rhabdomyomas are seen in approximately 60%-of children, but only 20%-of adults, and are usually asymptomatic. On MRI, they are well-defined masses on the ventricular septum, isointense to myocardium on T1, and hyperintense on T2. Although surgical excision is the mainstay of treatment, mTOR inhibitors can be used as a temporary treatment for symptomatic cardiac rhabdomyomas in TSC, especially in high risk and inoperable cases.[71]
About 20%-case of renal angiomyolipoma (AML) are associated with TSC. Presence of fat within AML can be demonstrated on CT, but best appreciated on T1 in- and opposed-phase images. Evaluation of lipid-poor AMLs can be a challenging and needs differentiation from RCC that is not uncommon in TSC. Multifocal and bilateral AMLs pose a challenge for surgical management, and thus RFA or cryoablation techniques are employed, especially for smaller tumors. mTOR inhibitors are used preoperatively that cause a significant reduction in size of lesions (38–95%) and reduce the risk of recurrence.[72]
The International Tuberous Sclerosis Complex Consensus Group reviewed the 2013 surveillance and management recommendations and published updated guidelines in 2021.[73] They have been summarized in [Fig. 10].
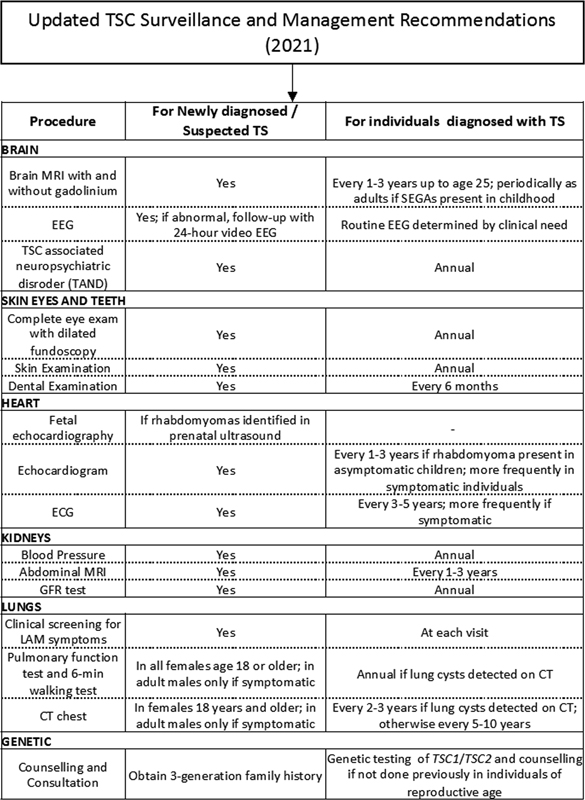
| fig 10:The updated International Tuberous Sclerosis Complex Consensus Group recommendations for surveillance and management. CT, computed tomography; ECG, electrocardiogram; EEG, electroencephalography; GFR, growth factor receptor; LAM, lymphangioleiomyomatosis; MRI, magnetic resonance imaging; SEGA, subependymal giant cell astrocytoma; TSC, tuberous sclerosis.
Conclusion
Identification and diagnosis of hereditary syndromes and genetic predisposition syndromes require an integrated approach of clinical evaluation, genetic testing, and radiological screening and surveillance. Surveillance recommendations have been developed for different HCSs over years based on the spectrum of associated cancers with the aim of prevention and early diagnosis. National and international collaboration to build cancer registries will be crucial for an evidenced-based and targeted approach to genetically predisposed individuals.
Conflict of Interest
None declared.
References
- Rutman AM, Kuo MD. Radiogenomics: creating a link between molecular diagnostics and diagnostic imaging. Eur J Radiol 2009; 70 (02) 232-241
- Garber JE, Offit K. Hereditary cancer predisposition syndromes. J Clin Oncol 2005; 23 (02) 276-292
- Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene 2004; 23 (38) 6445-6470
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65 (02) 87-108
- Rajkumar T, Soumittra N, Vidubala E. et al. Organization and running of the first comprehensive hereditary cancer clinic in India. Hered Cancer Clin Pract 2005; 3 (04) 165-170
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004; 10 (08) 789-799
- European Society of Radiology (ESR). Medical imaging in personalised medicine: a white paper of the research committee of the European Society of Radiology (ESR). Insights Imaging 2015; 6 (02) 141-155
- Kuhlen M, Borkhardt A. Cancer susceptibility syndromes in children in the area of broad clinical use of massive parallel sequencing. Eur J Pediatr 2015; 174 (08) 987-997
- Kuchenbaecker KB, Hopper JL, Barnes DR. et al; BRCA1 and BRCA2 Cohort Consortium. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017; 317 (23) 2402-2416
- Antoniou A, Pharoah PD, Narod S. et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72 (05) 1117-1130
- Mavaddat N, Barrowdale D, Andrulis IL. et al; HEBON, EMBRACE, GEMO Study Collaborators, kConFab Investigators, SWE-BRCA Collaborators, Consortium of Investigators of Modifiers of BRCA1/2. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol Biomarkers Prev 2012; 21 (01) 134-147
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Breast Cancer Linkage Consortium. Risks of cancer in BRCA1-mutation carriers. Lancet 1994; 343 (8899): 692-695
- Chen Y, Thompson W, Semenciw R, Mao Y. Epidemiology of contralateral breast cancer. Cancer Epidemiol Biomarkers Prev 1999; 8 (10) 855-861
- Graeser MK, Engel C, Rhiem K. et al. Contralateral breast cancer risk in BRCA1 and BRCA2 mutation carriers. J Clin Oncol 2009; 27 (35) 5887-5892
- Tai YC, Domchek S, Parmigiani G, Chen S. Breast cancer risk among male BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst 2007; 99 (23) 1811-1814
- Mitchell G, Antoniou AC, Warren R. et al. Mammographic density and breast cancer risk in BRCA1 and BRCA2 mutation carriers. Cancer Res 2006; 66 (03) 1866-1872
- Tice JA, Cummings SR, Ziv E, Kerlikowske K. Mammographic breast density and the Gail model for breast cancer risk prediction in a screening population. Breast Cancer Res Treat 2005; 94 (02) 115-122
- Wernli KJ, Ichikawa L, Kerlikowske K. et al. Surveillance breast MRI and mammography: comparison in women with a personal history of breast cancer. Radiology 2019; 292 (02) 311-318
- Saadatmand S, Geuzinge HA. et al. FaMRIsc study group. MRI versus mammography for breast cancer screening in women with familial risk (FaMRIsc): a multicentre, randomized, controlled trial. Lancet Oncol 2019; 20 (08) 1136-1147
- Raikhlin A, Curpen B, Warner E, Betel C, Wright B, Jong R. Breast MRI as an adjunct to mammography for breast cancer screening in high-risk patients: retrospective review. AJR Am J Roentgenol 2015; 204 (04) 889-897 Erratum in: AJR Am J Roentgenol. 2015 May;204(5):1137
- Girolimetti G, Perrone AM, Santini D. et al. BRCA-associated ovarian cancer: from molecular genetics to risk management. BioMed Res Int 2014; 2014: 787143
- Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res 2008; 68 (08) 2581-2586
- Bolton KL, Chenevix-Trench G, Goh C. et al; EMBRACE, kConFab Investigators, Cancer Genome Atlas Research Network. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 2012; 307 (04) 382-390
- Evans DG, Gaarenstroom KN, Stirling D. et al. Screening for familial ovarian cancer: poor survival of BRCA1/2 related cancers. J Med Genet 2009; 46 (09) 593-597
- Stirling D, Evans DG, Pichert G. et al. Screening for familial ovarian cancer: failure of current protocols to detect ovarian cancer at an early stage according to the international Federation of gynecology and obstetrics system. J Clin Oncol 2005; 23 (24) 5588-5596
- Toss A, Molinaro E, Sammarini M. et al. Hereditary ovarian cancers: state of the art. Minerva Med 2019; 110 (04) 301-319
- Bougeard G, Renaux-Petel M, Flaman JM. et al. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol 2015; 33 (21) 2345-2352
- Chompret A, Brugières L, Ronsin M. et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 2000; 82 (12) 1932-1937
- Bougeard G, Sesboüé R, Baert-Desurmont S. et al; French LFS working group. Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet 2008; 45 (08) 535-538
- Hendrickson PG, Luo Y, Kohlmann W, Schiffman J, Maese L, Bishop AJ, Lloyd S, Kokeny KE, Hitchcock YJ, Poppe MM, Gaffney DK, Tao R. Radiation therapy and secondary malignancy in Li-Fraumeni syndrome: A hereditary cancer registry study. Cancer Med 2020; Nov; 9 (21) 7954-7963 DOI: 10.1002/cam4.3427.
- Villani A, Shore A, Wasserman JD. et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol 2016; 17 (09) 1295-1305
- Hendrickson PG, Luo Y, Kohlmann W. et al. Radiation therapy and secondary malignancy in Li-Fraumeni syndrome: a hereditary cancer registry study. Cancer Med 2020; 9 (21) 7954-7963
- Nogueira ST, Lima EN, Nóbrega AF. et al. 18F-FDG PET-CT for surveillance of Brazilian patients with Li-Fraumeni syndrome. Front Oncol 2015; 5: 38
- Ballinger ML, Best A, Mai PL. et al. Baseline surveillance in li-fraumeni syndrome using whole-body magnetic resonance imaging: a meta-analysis. JAMA Oncol 2017; 3 (12) 1634-1639
- Villani A, Tabori U, Schiffman J. et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. Lancet Oncol 2011; 12 (06) 559-567
- O'Neill AF, Voss SD, Jagannathan JP. et al. Screening with whole-body magnetic resonance imaging in pediatric subjects with Li-Fraumeni syndrome: a single institution pilot study. Pediatr Blood Cancer 2018; 65 (02) e26822
- Tabori U, Shlien A, Baskin B. et al. TP53 alterations determine clinical subgroups and survival of patients with choroid plexus tumors. J Clin Oncol 2010; 28 (12) 1995-2001
- Orr BA, Clay MR, Pinto EM, Kesserwan C. An update on the central nervous system manifestations of Li-Fraumeni syndrome. Acta Neuropathol 2020; 139 (04) 669-687
- Frebourg T, Bajalica Lagercrantz S, Oliveira C, Magenheim R, Evans DG. European Reference Network GENTURIS. Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes. Eur J Hum Genet 2020; 28 (10) 1379-1386
- Farhadi F, Nikpanah M, Paschall AK. et al. Clear cell renal cell carcinoma growth correlates with baseline diffusion-weighted MRI in Von Hippel-Lindau disease. Radiology 2020; 295 (03) 583-590
- Ganeshan D, Menias CO, Pickhardt PJ. et al. Tumors in von Hippel-Lindau syndrome: from head to toe-comprehensive state-of-the-art review. Radiographics 2018; 38 (03) 849-866
- Jilg CA, Neumann HP, Gläsker S. et al. Nephron sparing surgery in von Hippel-Lindau associated renal cell carcinoma; clinicopathological long-term follow-up. Fam Cancer 2012; 11 (03) 387-394
- Filling-Katz MR, Choyke PL, Oldfield E. et al. Central nervous system involvement in Von Hippel-Lindau disease. Neurology 1991; 41 (01) 41-46
- Eisenhofer G, Lenders JW, Timmers H. et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem 2011; 57 (03) 411-420
- Mori H, Wakabayashi H, Saito S. et al. Comparison of detectability between WB-MRI and MIBG images in patients with metastatic pheochromocytoma and paraganglioma. J Nucl Med 2021; 62: 1111
- Charlesworth M, Verbeke CS, Falk GA, Walsh M, Smith AM, Morris-Stiff G. Pancreatic lesions in von Hippel-Lindau disease? A systematic review and meta-synthesis of the literature. J Gastrointest Surg 2012; 16 (07) 1422-1428
- Khanna L, Prasad SR, Sunnapwar A. et al. Pancreatic neuroendocrine neoplasms: 2020 update on pathologic and imaging findings and classification. Radiographics 2020; 40 (05) 1240-1262
- Schmid S, Gillessen S, Binet I. et al. Management of von Hippel-Lindau disease: an interdisciplinary review. Oncol Res Treat 2014; 37 (12) 761-771
- Binderup ML, Bisgaard ML, Harbud V. et al; Danish vHL Coordination Group. Von Hippel-Lindau disease (vHL). National clinical guideline for diagnosis and surveillance in Denmark. 3rd edition. Dan Med J 2013; 60 (12) B4763
- Kruizinga RC, Sluiter WJ, de Vries EG. et al. Calculating optimal surveillance for detection of von Hippel-Lindau-related manifestations. Endocr Relat Cancer 2013; 21 (01) 63-71
- VHL Family Alliance. The VHL handbook: what you need to know about VHL—a reference handbook for people with von Hippel-Lindau disease, their families, and support personnel. 2020 https://www.vhl.org/care-treatment/resources/
- Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol 2014; 386 (1-2): 2-15
- Kamilaris CDC, Stratakis CA. Multiple Endocrine Neoplasia Type 1 (MEN1): An Update and the Significance of Early Genetic and Clinical Diagnosis. Front Endocrinol (Lausanne) 2019; Jun 11: 10-339 DOI: 10.3389/fendo.2019.00339.
- Moline J, Eng C. Multiple endocrine neoplasia type 2: an overview. Genet Med 2011; 13 (09) 755-764
- Wells Jr SA, Asa SL, Dralle H. et al; American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015; 25 (06) 567-610
- Eichhorn-Wharry LI, Carlin AM, Talpos GB. Mild hypercalcemia: an indication to select 4-dimensional computed tomography scan for preoperative localization of parathyroid adenomas. Am J Surg 2011; 201 (03) 334-338 , discussion 338
- Nael K, Hur J, Bauer A. et al. Dynamic 4D MRI for characterization of parathyroid adenomas: multi- parametric analysis. AJNR Am J Neuroradiol 2015; 36 (11) 2147-2152
- Niederle B, Selberherr A, Bartsch DK. et al. Multiple endocrine neoplasia type 1 and the pancreas: diagnosis and treatment of functioning and non-functioning pancreatic and duodenal neuroendocrine neoplasia within the MEN1 syndrome - an International Consensus Statement. Neuroendocrinology 2021; 111 (07) 609-630
- Grajo JR, Paspulati RM, Sahani DV, Kambadakone A. Multiple endocrine neoplasia syndromes: a comprehensive imaging review. Radiol Clin North Am 2016; 54 (03) 441-451
- Iglesias P, Cardona J, Díez JJ. The pituitary in nuclear medicine imaging. Eur J Intern Med 2019; 68: 6-12
- Scarsbrook AF, Thakker RV, Wass JAH, et al. Multiple endocrine neoplasia: spectrum of radiologic appearances and discussion of a multitechnique imaging approach. Radiographics 2006; 26: 433-451
- Castinetti F, Taïeb D. Positron emission tomography imaging in medullary thyroid carcinoma: time for reappraisal?. Thyroid 2021; 31 (02) 151-155
- Gagel RF, Tashjian Jr AH, Cummings T. et al. The clinical outcome of prospective screening for multiple endocrine neoplasia type 2a. An 18-year experience. N Engl J Med 1988; 318 (08) 478-484
- de Laat JM, van Leeuwaarde RS, Valk GD. The importance of an early and accurate MEN1 diagnosis. Front Endocrinol (Lausanne) 2018; 9: 533
- Peters JM, Prohl AK, Tomas-Fernandez XK. et al. Tubers are neither static nor discrete: evidence from serial diffusion tensor imaging. Neurology 2015; 85 (18) 1536-1545
- Pollock JM, Whitlow CT, Tan H, Kraft RA, Burdette JH, Maldjian JA. Pulsed arterial spin-labeled MR imaging evaluation of tuberous sclerosis. AJNR Am J Neuroradiol 2009; 30 (04) 815-820
- Liu S, Cai Y, Rong R. et al. Tuberous sclerosis complex (TSC) with epilepsy on 18F-FDG simultaneous PET/MR. Eur J Nucl Med Mol Imaging 2020; 47 (10) 2471-2472
- Ruppe V, Dilsiz P, Reiss CS. et al. Developmental brain abnormalities in tuberous sclerosis complex: a comparative tissue analysis of cortical tubers and perituberal cortex. Epilepsia 2014; 55 (04) 539-550
- Roth J, Roach ES, Bartels U. et al. Subependymal giant cell astrocytoma: diagnosis, screening, and treatment. Recommendations from the International Tuberous Sclerosis Complex Consensus Conference 2012. Pediatr Neurol 2013; 49 (06) 439-444
- Jóźwiak S, Nabbout R, Curatolo P. participants of the TSC Consensus Meeting for SEGA and Epilepsy Management. Management of subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis complex (TSC): Clinical recommendations. Eur J Paediatr Neurol 2013; 17 (04) 348-352
- Sugalska M, Tomik A, Jóźwiak S, Werner B. Treatment of cardiac rhabdomyomas with mTOR inhibitors in children with tuberous sclerosis complex-a systematic review. Int J Environ Res Public Health 2021; 18 (09) 4907
- Staehler M, Sauter M, Helck A. et al. Nephron-sparing resection of angiomyolipoma after sirolimus pretreatment in patients with tuberous sclerosis. Int Urol Nephrol 2012; 44 (06) 1657-1661
- Northrup H, Aronow ME, Bebin EM. et al; International Tuberous Sclerosis Complex Consensus Group. Updated International Tuberous Sclerosis Complex Diagnostic Criteria and Surveillance and Management Recommendations. Pediatr Neurol 2021; 123: 50-66
| Fig 1 :Imaging and clinical screening recommendations in BRCA carriers. CT, computed tomography; HBOCS, hereditary breast and ovarian cancer syndrome; MRI, magnetic resonance imaging; RRSO, risk-reducing salpingo-oophorectomy.
| Figure 2:Classic and Chompret criteria for diagnosis of Li-Fraumeni syndrome (LFS).
| Figure 3:Surveillance recommendations in carriers of germline disease-causing TP53 variants. LFS, Li-Fraumeni syndrome; MRI, magnetic resonance imaging.
| Figure 4:Individuals who should be tested for VHL mutation. CNS, central nervous system; Hb, hemoglobin; PNET,—; RCC, renal cell carcinoma; VHL, von Hippel-Lindau disease.
| Figure 5:VHL alliance suggested surveillance guidelines. MRI, magnetic resonance imaging; VHL, von Hippel-Lindau disease.
| Figure 6:Associated abnormalities with MEN syndromes. MEN, multiple endocrine neoplasia syndrome; MTC, medullary thyroid cancer.
| Fig 7:Guidelines for screening protocol for MEN1 syndrome. CT, computed tomography; EUS, endoscopic ultrasound; IGF-1, insulin-like growth factor-1; MRI, magnetic resonance imaging; MEN1, multiple endocrine neoplasia 1; NET, neuroendocrine tumor; PTH, parathormone; VIP, vasoactive intestinal peptide.
| fig 8:Guidelines for screening protocol for MEN2 syndrome. MEN2, multiple endocrine neoplasia 2; MTC, medullary thyroid cancer.
| fig 9:Clinical features for diagnosis of TSC. CT, computed tomography; SEGA, subependymal giant cell astrocytoma; TSC, tuberous sclerosis.
| fig 10:The updated International Tuberous Sclerosis Complex Consensus Group recommendations for surveillance and management. CT, computed tomography; ECG, electrocardiogram; EEG, electroencephalography; GFR, growth factor receptor; LAM, lymphangioleiomyomatosis; MRI, magnetic resonance imaging; SEGA, subependymal giant cell astrocytoma; TSC, tuberous sclerosis.
References
- Rutman AM, Kuo MD. Radiogenomics: creating a link between molecular diagnostics and diagnostic imaging. Eur J Radiol 2009; 70 (02) 232-241
- Garber JE, Offit K. Hereditary cancer predisposition syndromes. J Clin Oncol 2005; 23 (02) 276-292
- Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene 2004; 23 (38) 6445-6470
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65 (02) 87-108
- Rajkumar T, Soumittra N, Vidubala E. et al. Organization and running of the first comprehensive hereditary cancer clinic in India. Hered Cancer Clin Pract 2005; 3 (04) 165-170
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004; 10 (08) 789-799
- European Society of Radiology (ESR). Medical imaging in personalised medicine: a white paper of the research committee of the European Society of Radiology (ESR). Insights Imaging 2015; 6 (02) 141-155
- Kuhlen M, Borkhardt A. Cancer susceptibility syndromes in children in the area of broad clinical use of massive parallel sequencing. Eur J Pediatr 2015; 174 (08) 987-997
- Kuchenbaecker KB, Hopper JL, Barnes DR. et al; BRCA1 and BRCA2 Cohort Consortium. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017; 317 (23) 2402-2416
- Antoniou A, Pharoah PD, Narod S. et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72 (05) 1117-1130
- Mavaddat N, Barrowdale D, Andrulis IL. et al; HEBON, EMBRACE, GEMO Study Collaborators, kConFab Investigators, SWE-BRCA Collaborators, Consortium of Investigators of Modifiers of BRCA1/2. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol Biomarkers Prev 2012; 21 (01) 134-147
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Breast Cancer Linkage Consortium. Risks of cancer in BRCA1-mutation carriers. Lancet 1994; 343 (8899): 692-695
- Chen Y, Thompson W, Semenciw R, Mao Y. Epidemiology of contralateral breast cancer. Cancer Epidemiol Biomarkers Prev 1999; 8 (10) 855-861
- Graeser MK, Engel C, Rhiem K. et al. Contralateral breast cancer risk in BRCA1 and BRCA2 mutation carriers. J Clin Oncol 2009; 27 (35) 5887-5892
- Tai YC, Domchek S, Parmigiani G, Chen S. Breast cancer risk among male BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst 2007; 99 (23) 1811-1814
- Mitchell G, Antoniou AC, Warren R. et al. Mammographic density and breast cancer risk in BRCA1 and BRCA2 mutation carriers. Cancer Res 2006; 66 (03) 1866-1872
- Tice JA, Cummings SR, Ziv E, Kerlikowske K. Mammographic breast density and the Gail model for breast cancer risk prediction in a screening population. Breast Cancer Res Treat 2005; 94 (02) 115-122
- Wernli KJ, Ichikawa L, Kerlikowske K. et al. Surveillance breast MRI and mammography: comparison in women with a personal history of breast cancer. Radiology 2019; 292 (02) 311-318
- Saadatmand S, Geuzinge HA. et al. FaMRIsc study group. MRI versus mammography for breast cancer screening in women with familial risk (FaMRIsc): a multicentre, randomized, controlled trial. Lancet Oncol 2019; 20 (08) 1136-1147
- Raikhlin A, Curpen B, Warner E, Betel C, Wright B, Jong R. Breast MRI as an adjunct to mammography for breast cancer screening in high-risk patients: retrospective review. AJR Am J Roentgenol 2015; 204 (04) 889-897 Erratum in: AJR Am J Roentgenol. 2015 May;204(5):1137
- Girolimetti G, Perrone AM, Santini D. et al. BRCA-associated ovarian cancer: from molecular genetics to risk management. BioMed Res Int 2014; 2014: 787143
- Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res 2008; 68 (08) 2581-2586
- Bolton KL, Chenevix-Trench G, Goh C. et al; EMBRACE, kConFab Investigators, Cancer Genome Atlas Research Network. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 2012; 307 (04) 382-390
- Evans DG, Gaarenstroom KN, Stirling D. et al. Screening for familial ovarian cancer: poor survival of BRCA1/2 related cancers. J Med Genet 2009; 46 (09) 593-597
- Stirling D, Evans DG, Pichert G. et al. Screening for familial ovarian cancer: failure of current protocols to detect ovarian cancer at an early stage according to the international Federation of gynecology and obstetrics system. J Clin Oncol 2005; 23 (24) 5588-5596
- Toss A, Molinaro E, Sammarini M. et al. Hereditary ovarian cancers: state of the art. Minerva Med 2019; 110 (04) 301-319
- Bougeard G, Renaux-Petel M, Flaman JM. et al. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol 2015; 33 (21) 2345-2352
- Chompret A, Brugières L, Ronsin M. et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 2000; 82 (12) 1932-1937
- Bougeard G, Sesboüé R, Baert-Desurmont S. et al; French LFS working group. Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet 2008; 45 (08) 535-538
- Hendrickson PG, Luo Y, Kohlmann W, Schiffman J, Maese L, Bishop AJ, Lloyd S, Kokeny KE, Hitchcock YJ, Poppe MM, Gaffney DK, Tao R. Radiation therapy and secondary malignancy in Li-Fraumeni syndrome: A hereditary cancer registry study. Cancer Med 2020; Nov; 9 (21) 7954-7963 DOI: 10.1002/cam4.3427.
- Villani A, Shore A, Wasserman JD. et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol 2016; 17 (09) 1295-1305
- Hendrickson PG, Luo Y, Kohlmann W. et al. Radiation therapy and secondary malignancy in Li-Fraumeni syndrome: a hereditary cancer registry study. Cancer Med 2020; 9 (21) 7954-7963
- Nogueira ST, Lima EN, Nóbrega AF. et al. 18F-FDG PET-CT for surveillance of Brazilian patients with Li-Fraumeni syndrome. Front Oncol 2015; 5: 38
- Ballinger ML, Best A, Mai PL. et al. Baseline surveillance in li-fraumeni syndrome using whole-body magnetic resonance imaging: a meta-analysis. JAMA Oncol 2017; 3 (12) 1634-1639
- Villani A, Tabori U, Schiffman J. et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. Lancet Oncol 2011; 12 (06) 559-567
- O'Neill AF, Voss SD, Jagannathan JP. et al. Screening with whole-body magnetic resonance imaging in pediatric subjects with Li-Fraumeni syndrome: a single institution pilot study. Pediatr Blood Cancer 2018; 65 (02) e26822
- Tabori U, Shlien A, Baskin B. et al. TP53 alterations determine clinical subgroups and survival of patients with choroid plexus tumors. J Clin Oncol 2010; 28 (12) 1995-2001
- Orr BA, Clay MR, Pinto EM, Kesserwan C. An update on the central nervous system manifestations of Li-Fraumeni syndrome. Acta Neuropathol 2020; 139 (04) 669-687
- Frebourg T, Bajalica Lagercrantz S, Oliveira C, Magenheim R, Evans DG. European Reference Network GENTURIS. Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes. Eur J Hum Genet 2020; 28 (10) 1379-1386
- Farhadi F, Nikpanah M, Paschall AK. et al. Clear cell renal cell carcinoma growth correlates with baseline diffusion-weighted MRI in Von Hippel-Lindau disease. Radiology 2020; 295 (03) 583-590
- Ganeshan D, Menias CO, Pickhardt PJ. et al. Tumors in von Hippel-Lindau syndrome: from head to toe-comprehensive state-of-the-art review. Radiographics 2018; 38 (03) 849-866
- Jilg CA, Neumann HP, Gläsker S. et al. Nephron sparing surgery in von Hippel-Lindau associated renal cell carcinoma; clinicopathological long-term follow-up. Fam Cancer 2012; 11 (03) 387-394
- Filling-Katz MR, Choyke PL, Oldfield E. et al. Central nervous system involvement in Von Hippel-Lindau disease. Neurology 1991; 41 (01) 41-46
- Eisenhofer G, Lenders JW, Timmers H. et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem 2011; 57 (03) 411-420
- Mori H, Wakabayashi H, Saito S. et al. Comparison of detectability between WB-MRI and MIBG images in patients with metastatic pheochromocytoma and paraganglioma. J Nucl Med 2021; 62: 1111
- Charlesworth M, Verbeke CS, Falk GA, Walsh M, Smith AM, Morris-Stiff G. Pancreatic lesions in von Hippel-Lindau disease? A systematic review and meta-synthesis of the literature. J Gastrointest Surg 2012; 16 (07) 1422-1428
- Khanna L, Prasad SR, Sunnapwar A. et al. Pancreatic neuroendocrine neoplasms: 2020 update on pathologic and imaging findings and classification. Radiographics 2020; 40 (05) 1240-1262
- Schmid S, Gillessen S, Binet I. et al. Management of von Hippel-Lindau disease: an interdisciplinary review. Oncol Res Treat 2014; 37 (12) 761-771
- Binderup ML, Bisgaard ML, Harbud V. et al; Danish vHL Coordination Group. Von Hippel-Lindau disease (vHL). National clinical guideline for diagnosis and surveillance in Denmark. 3rd edition. Dan Med J 2013; 60 (12) B4763
- Kruizinga RC, Sluiter WJ, de Vries EG. et al. Calculating optimal surveillance for detection of von Hippel-Lindau-related manifestations. Endocr Relat Cancer 2013; 21 (01) 63-71
- VHL Family Alliance. The VHL handbook: what you need to know about VHL—a reference handbook for people with von Hippel-Lindau disease, their families, and support personnel. 2020 https://www.vhl.org/care-treatment/resources/
- Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol 2014; 386 (1-2): 2-15
- Kamilaris CDC, Stratakis CA. Multiple Endocrine Neoplasia Type 1 (MEN1): An Update and the Significance of Early Genetic and Clinical Diagnosis. Front Endocrinol (Lausanne) 2019; Jun 11: 10-339 DOI: 10.3389/fendo.2019.00339.
- Moline J, Eng C. Multiple endocrine neoplasia type 2: an overview. Genet Med 2011; 13 (09) 755-764
- Wells Jr SA, Asa SL, Dralle H. et al; American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015; 25 (06) 567-610
- Eichhorn-Wharry LI, Carlin AM, Talpos GB. Mild hypercalcemia: an indication to select 4-dimensional computed tomography scan for preoperative localization of parathyroid adenomas. Am J Surg 2011; 201 (03) 334-338 , discussion 338
- Nael K, Hur J, Bauer A. et al. Dynamic 4D MRI for characterization of parathyroid adenomas: multi- parametric analysis. AJNR Am J Neuroradiol 2015; 36 (11) 2147-2152
- Niederle B, Selberherr A, Bartsch DK. et al. Multiple endocrine neoplasia type 1 and the pancreas: diagnosis and treatment of functioning and non-functioning pancreatic and duodenal neuroendocrine neoplasia within the MEN1 syndrome - an International Consensus Statement. Neuroendocrinology 2021; 111 (07) 609-630
- Grajo JR, Paspulati RM, Sahani DV, Kambadakone A. Multiple endocrine neoplasia syndromes: a comprehensive imaging review. Radiol Clin North Am 2016; 54 (03) 441-451
- Iglesias P, Cardona J, Díez JJ. The pituitary in nuclear medicine imaging. Eur J Intern Med 2019; 68: 6-12
- Scarsbrook AF, Thakker RV, Wass JAH, et al. Multiple endocrine neoplasia: spectrum of radiologic appearances and discussion of a multitechnique imaging approach. Radiographics 2006; 26: 433-451
- Castinetti F, Taïeb D. Positron emission tomography imaging in medullary thyroid carcinoma: time for reappraisal?. Thyroid 2021; 31 (02) 151-155
- Gagel RF, Tashjian Jr AH, Cummings T. et al. The clinical outcome of prospective screening for multiple endocrine neoplasia type 2a. An 18-year experience. N Engl J Med 1988; 318 (08) 478-484
- de Laat JM, van Leeuwaarde RS, Valk GD. The importance of an early and accurate MEN1 diagnosis. Front Endocrinol (Lausanne) 2018; 9: 533
- Peters JM, Prohl AK, Tomas-Fernandez XK. et al. Tubers are neither static nor discrete: evidence from serial diffusion tensor imaging. Neurology 2015; 85 (18) 1536-1545
- Pollock JM, Whitlow CT, Tan H, Kraft RA, Burdette JH, Maldjian JA. Pulsed arterial spin-labeled MR imaging evaluation of tuberous sclerosis. AJNR Am J Neuroradiol 2009; 30 (04) 815-820
- Liu S, Cai Y, Rong R. et al. Tuberous sclerosis complex (TSC) with epilepsy on 18F-FDG simultaneous PET/MR. Eur J Nucl Med Mol Imaging 2020; 47 (10) 2471-2472
- Ruppe V, Dilsiz P, Reiss CS. et al. Developmental brain abnormalities in tuberous sclerosis complex: a comparative tissue analysis of cortical tubers and perituberal cortex. Epilepsia 2014; 55 (04) 539-550
- Roth J, Roach ES, Bartels U. et al. Subependymal giant cell astrocytoma: diagnosis, screening, and treatment. Recommendations from the International Tuberous Sclerosis Complex Consensus Conference 2012. Pediatr Neurol 2013; 49 (06) 439-444
- Jóźwiak S, Nabbout R, Curatolo P. participants of the TSC Consensus Meeting for SEGA and Epilepsy Management. Management of subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis complex (TSC): Clinical recommendations. Eur J Paediatr Neurol 2013; 17 (04) 348-352
- Sugalska M, Tomik A, Jóźwiak S, Werner B. Treatment of cardiac rhabdomyomas with mTOR inhibitors in children with tuberous sclerosis complex-a systematic review. Int J Environ Res Public Health 2021; 18 (09) 4907
- Staehler M, Sauter M, Helck A. et al. Nephron-sparing resection of angiomyolipoma after sirolimus pretreatment in patients with tuberous sclerosis. Int Urol Nephrol 2012; 44 (06) 1657-1661
- Northrup H, Aronow ME, Bebin EM. et al; International Tuberous Sclerosis Complex Consensus Group. Updated International Tuberous Sclerosis Complex Diagnostic Criteria and Surveillance and Management Recommendations. Pediatr Neurol 2021; 123: 50-66