 PDF
PDF  Views
Views  Share
Share


Imaging spectrum of primary malignant renal neoplasms in children
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2012; 33(04): 242-249
DOI: DOI: 10.4103/0971-5851.107107
Abstract
Wilms′ tumor (WT) is the most common abdominal tumor in children. Many pediatric renal tumors in the past were categorized as WT; however, in recent years, several specific renal tumors have been recognized as distinct pathological entities. The age and clinical presentation of the child and distinctive imaging features may help in reaching a specific diagnosis in most cases. This is important as it has implications on the pre-operative diagnostic work-up and prognosis of the child. However, it is often not possible to differentiate one from the other pediatric renal tumor on the basis of imaging alone, and the final diagnosis is often made at histological examination of the surgical specimen. This article reviews the imaging features of primary malignant renal neoplasms in children along with their clinical presentation and pathological features.
Publication History
Article published online:
20 July 2021
© 2012. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Wilms’ tumor (WT) is the most common abdominal tumor in children. Many pediatric renal tumors in the past were categorized as WT; however, in recent years, several specific renal tumors have been recognized as distinct pathological entities. The age and clinical presentation of the child and distinctive imaging features may help in reaching a specific diagnosis in most cases. This is important as it has implications on the pre-operative diagnostic work-up and prognosis of the child. However, it is often not possible to differentiate one from the other pediatric renal tumor on the basis of imaging alone, and the final diagnosis is often made at histological examination of the surgical specimen. This article reviews the imaging features of primary malignant renal neoplasms in children along with their clinical presentation and pathological features.
INTRODUCTION
The diagnosis of a renal neoplasm in children is suggested by clinical history, age at presentation and distinctive imaging features. In a neonate, most renal masses are due to developmental abnormalities like congenital hydronephrosis or multicystic dysplastic kidney. Beyond the first year of life and during the first decade, primary tumors of the kidney are more common.[1] Primary renal neoplasms of childhood are classified as low-, intermediate- and high-risk tumors [Table 1].
Table 1
Renal tumors of childhood (Revised International Society of Paediatric Oncology [SIOP] working classification of renal tumors of childhood)

DISCUSSION
Wilms’ tumor
Wilms’ tumor (WT)/nephroblastoma is the most common pediatric renal neoplasm, accounting for 87% of pediatric renal masses. The peak incidence is between 3 and 4 years of age; however bilateral tumors (4–13% cases) present at a younger age. They are usually synchronous and have a higher incidence of nephrogenic rests and associated congenital anomalies like cryptoorchidism, hemihypertrophy, hypospadias and sporadic aniridia.[2,3] WTs are usually sporadic, with familial tumors seen in approximately 1% of the cases. Specific germline mutations on WT1 and WT2 genes located on chromosome 11 may be seen in a minority of the cases.[2,4]
Grossly, WT is a solid tumor with a fibrous pseudocapsule, variable foci of hemorrhage and necrosis. The classic WT is triphasic, with variable amounts of blastema, stroma and epithelium [Figure 1]. Presence of additional tissue not normally found in the kidney (like bone, cartilage or muscle) makes it a teratoid tumor. Prognosis depends on degree of anaplasia, and anaplastic change denotes unfavourable histology. Majority (~90%) of WTs have favourable histology.[1,2]
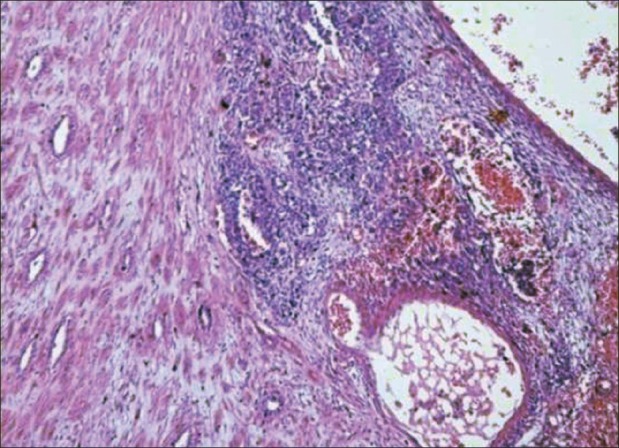
| Fig. 1 Photomicrograph showing triphasic nephroblastoma with blastemal, stromal and epithelial components
Clinically, the child usually presents with a painless abdominal mass. Hypertension due to renin production by the tumor and microscopic hematuria are seen in about 25% of the cases. Gross hematuria is very uncommon, and signifies collecting system invasion.[2,5]
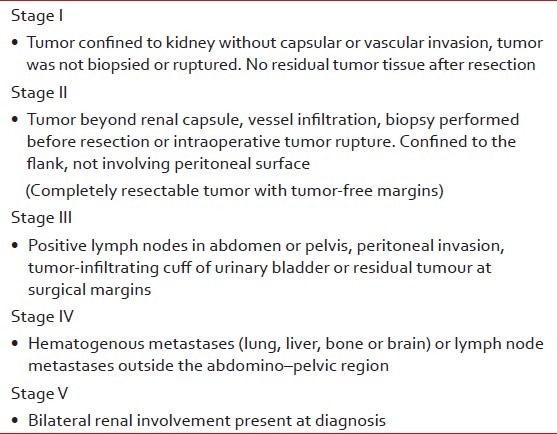
Staging of WT [Table 2] is determined by imaging studies and the surgical and pathologic findings at nephrectomy. Ultrasound is the first imaging modality as for any child with abdominal mass. X-ray chest is done at baseline as lung is the most common site for metastases.[4,6,7] On ultrasonography (USG), WT is seen as a heteroechoic mass with variable foci of hemorrhage, necrosis and calcification [Figure 2]. Renal vein/inferior vena cava (IVC)/hepatic veins should be assessed for tumor thrombus. Caval extension of tumor thrombus is seen in about 4% of the cases, with the thrombus extending into the right atrium [Figure 3], and such patients benefit from pre-surgical chemotherapy. Contralateral kidney must always be inspected for its absence, synchronous tumor, nephrogenic rests and congenital abnormalities as it affects the management [Figure 4].[1,2,6]
Table 2
Wilms’ tumor – clinicopathological – National Wilms’ Tumor Study (NWTS) staging
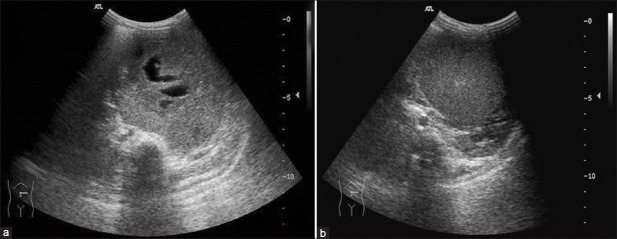
| Fig. 2(a and b) Wilms’ tumor – transverse ultrasonography shows solid mass with anechoic necrotic areas and echogenic pseudocapsule
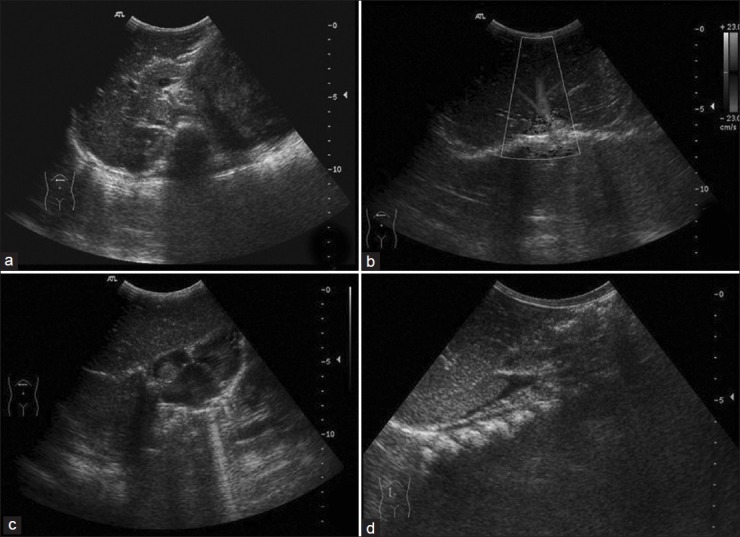
| Fig. 3 Ultrasonography showing caval thrombus with extension into the right atrium in a case of Wilms’ tumor
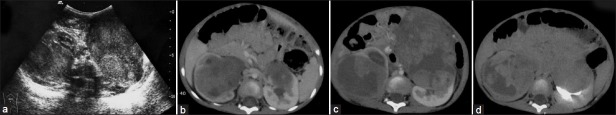
| Fig. 4 Stage V Wilms’ tumor in a 2-year-old child – bilateral heterodense renal masses showing extensive necrosis and calcification. Right renal mass is extending into pelvicalyceal system. Bilateral renal vessels are normal. Computed tomography urography image shows excretion from left kidney. Pre-operative chemotherapy followed by right nephroureterectomy and left partial nephrectomy was done. It was Stage III tumor on the right side and Stage II tumor on the left side
Abdominal computed tomography (CT) is more sensitive than USG in identifying the tumor extent and nodal as well as liver involvement.[1] With the advent of multidetector CT, multiplanar reformatting (MPR) and maximum intensity projection (MIP) images allow viewing the mass in entirety and one can clearly visualize regional invasion and the relationship of the vessels with the mass. On non-contrast computed tomography (NCCT), it is seen as a well-circumscribed heterodense mass with areas of low attenuation due to necrosis, fat and old hemorrhage. Acute hemorrhage shows fluid–fluid level and calcification is seen in ~9% of the cases. On contrast-enhanced computed tomography (CECT), tumor enhances less than normal renal parenchyma. A claw or beak sign is seen due to the normal renal tissue displaced by tumor [Figure 5]. CECT is essential for assessment of lymphadenopathy, vascular invasion, metastases to liver, peritoneal invasion [Figures [Figures66–8] and contralateral kidney for presence of synchronous small WT and nephrogenic rests. Magnetic resonance imaging (MRI) has been reported as the most sensitive modality for determination of caval patency, but it requires sedation, general anesthesia and long scanning time. The disadvantage of CT is of course the radiation burden, which should be kept as low as possible in pediatric patients.[1,2,5,6]
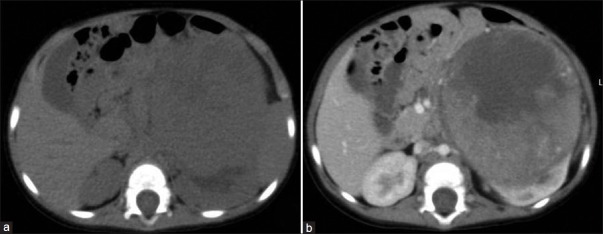
| Fig. 5 Stage I Wilms’ tumor (WT) – non-contrast (NCCT) and contrast-enhanced CT (CECT) images show a large well-encapsulated left lower pole renal mass with hypodense areas on NCCT and heterogeneous enhancement on post-contrast image. Claw sign – characteristic of WT is also seen. No vascular invasion is evident. Mass enhances less than normal renal parenchyma. It was a completely resectable tumor with tumour-free margins
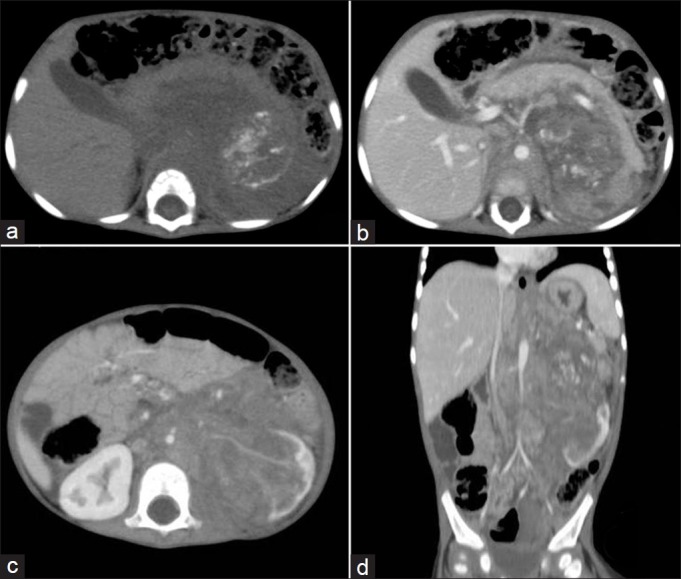
| Fig. 6 Stage III Wilms’ tumor – contrast-enhanced computed tomography (CECT) showing large heterogeneously enhancing mass with calcification, vascular encasement, periportal and retroperitoneal lymphadenopathy and pancreatic involvement. Multiplanar reconstruction and maximum intensity projection image shows the entire extent of mass extending till the diaphragm and spleen. The tumor is severely encasing the vessels and is densely adherent to diaphragm and pancreas; hence, it is non-resectable
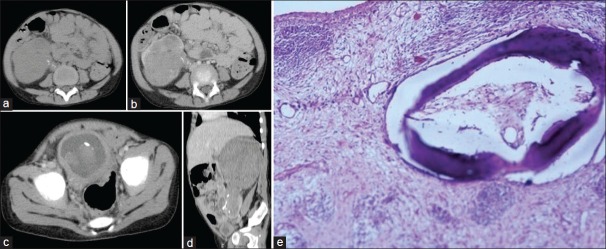
| Fig. 8 (a–c) Stage III Wilms’ tumor in a 3-year-old child with hematuria. Heterogeneous mass with calcification, extending into the pelvicalyceal system and ureter with tumor embolus in urinary bladder. Extension to ureter is better appreciated on multiplanar reformatting (MPR) image (d). Photomicrograph (e) showing teratoid tumor with focal areas of osteoid, hyaline and smooth muscle differentiation
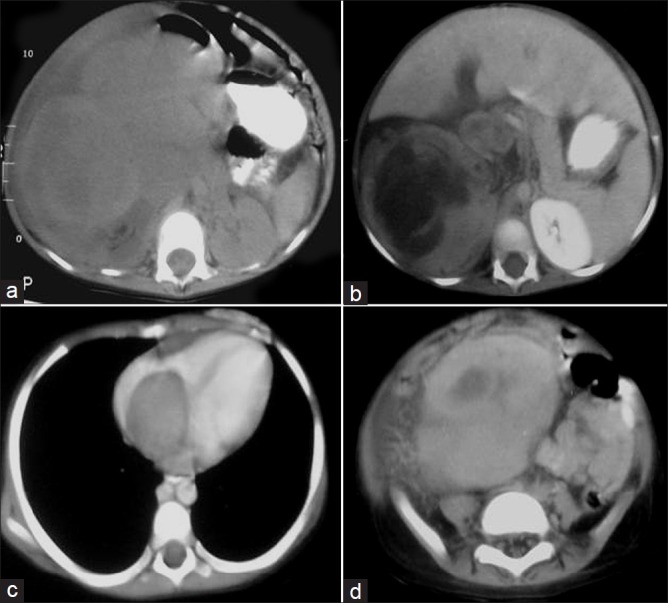
| Fig. 7 Stage III Wilms’ tumor – large lobulated heterogeneously enhancing right renal mass with renal vein and IVC thrombus extending into right atrium. Retroperitoneal lymphadenopathy, ascites and peritoneal involvement are seen
Lung is the most common site for metastases in WT and, traditionally, X-ray chest has been the means for assessing pulmonary metastases. CT chest has proved to be more sensitive for detection of pulmonary nodules [Figure 9]. However, there is controversy regarding treatment of CT-only lesions in the lung (not detected on chest X-ray) as there is no difference in survival statistics when a child is upstaged on the basis of CT findings.[1,6]
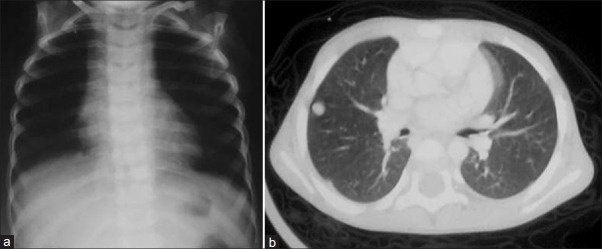
| Fig. 9 Chest X-ray and computed tomography scan of a case of Stage IV Wilms’ tumor – pulmonary nodules are not seen on X-ray chest
Nephroblastomatosis
Nephroblastomatosis consist of diffuse or multifocal involvement of kidney by nephrogenic rests (NRs), which are the foci of metanephric blastema that persist beyond 36 weeks gestation (i.e. when nephrogenesis is complete). The cause for these NRs could be incomplete induction of metanephric blastema to mature renal parenchyma by ureteric bud.[5] Incidence of NRs is ~1% in perinatal autopsies, and they have a potential for malignant transformation to WT. NRs have been identified in ~41% of the patients with unilateral WT and in ~94% and ~99% of metachronous and synchronous bilateral tumors, respectively [Figure 10].[8,9]
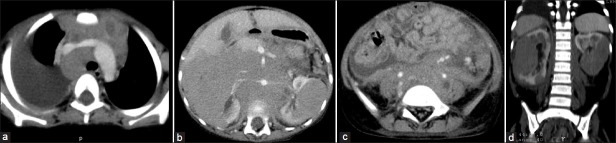
| Fig. 10 Nephroblastomatosis – progression to bilateral Wilms’ tumors. Right pleural effusion, mediastinal lymphadenopathy, bilateral renal masses, retroperitoneal lymphadenopathy and ascites are seen
NRs can be classified as perilobar (peripheral cortex or columns of Bertin) and intralobar on the basis of location and the syndromes with which they are associated. Intralobar NRs have a higher association with WT development.[5,10] NRs show variable proportions of blastema, stromal and epithelial elements. Based on histology, these are classified as dormant, sclerosing, hyperplastic or neoplastic.[8] Dormant and sclerosing NRs are usually microscopic, with no malignant potential; hyperplastic and neoplastic rests are larger, mitotically active and may show cellular atypia.[5,8]
Ultrasound is a less-sensitive modality than CT and MRI to detect macroscopic NRs.[1,3] The affected kidney may be enlarged and have indistinct corticomedullary junction. Kidney may be diffusely hypoechoic or may show focal hypoechoic nodules [Figures [Figures1111 and and12],12], making it difficult to differentiate from renal involvement in leukemia or lymphoma.[1–3] On CT, macroscopic NRs appear as low-attenuation peripheral nodules with poor enhancement relative to adjacent normal renal parenchyma [Figure 11d].[2] On MRI, these lesions appear as low signal intensity foci on both T1W and T2W images. These are rendered more conspicuous on contrast-enhanced MRI as they stand out against the normal enhancing parenchyma.[1–3,10]
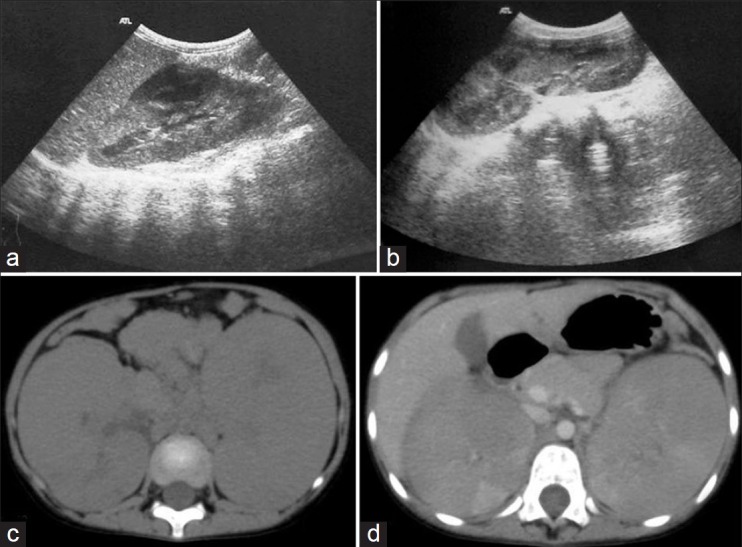
| Fig. 11 Nephroblastomatosis – ultrasonography (USG) and computed tomography images of a 4-year-old female showing bilateral enlarged kidneys with lobulated contour. Nephrogenic rests are seen as hypoechoic lesions on USG and as hypodense, non-enhancing lesions on contrast-enhanced computed tomography (CECT). They are better appreciated on CECT
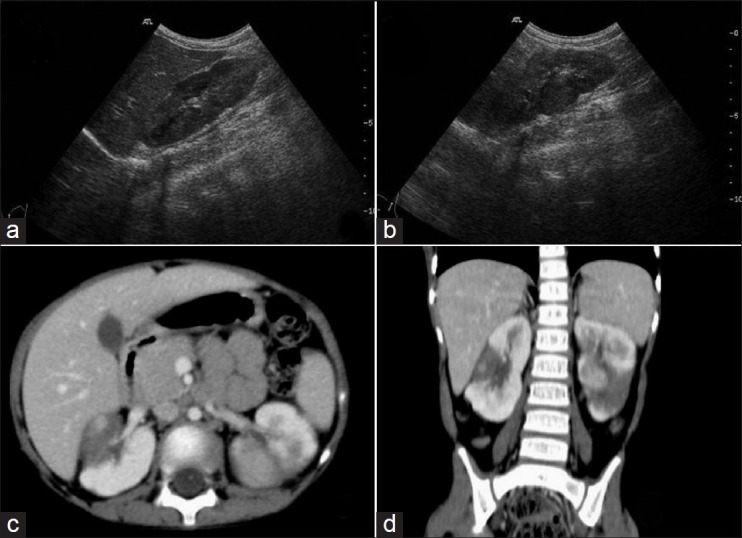
| Fig. 12 Nephroblastomatosis – after a 2-month interval, spontaneous regression of some lesions is evident. Renal size is decreased bilaterally and hypodense lesions (nephrogenic rests) are better appreciated
Clear cell sarcoma kidney
Clear cell sarcoma kidney (CCSK) is a distinct entity accounting for 4% of all childhood renal neoplasms.[7] It was formerly considered as a highly aggressive variant of WT. It has a strong male preponderance, with peak incidence at 1–4 years of age.[1,2]
There are no specific radiological features to help distinguish CCSK from WT [Figure 13]. It is characterized by aggressive behavior and a high rate of relapse and mortality than WT. There is a strong propensity for skeletal metastases (over 20% risk); bone scintigraphy is indicated for staging purposes once this lesion is diagnosed. Other sites of metastases are lung, liver, lymph nodes and brain.[2,7]
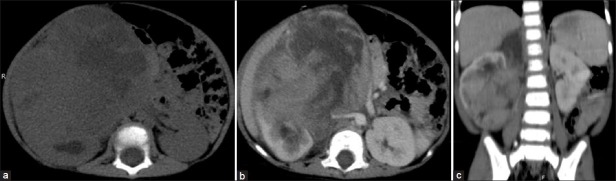
| Fig. 13 Clear cell sarcoma kidney – heterogeneously enhancing right renal mass with extensive necrosis
Rhabdoid tumor
It is the most aggressive malignant neoplasm of kidney in children, and accounts for about 2% of pediatric renal malignancies. Most cases are diagnosed in the first year of life, with a slight male preponderance [1.5:1].[2,7,11] Child may present with hematuria or symptoms referable to metastatic disease. Its name is derived from its histological appearance, which resembles that of a tumor of skeletal muscle origin, although a myogenic origin has not been proved.[2]
Imaging demonstrates a large, centrally located, heterogeneous soft tissue mass involving the renal hilum with indistinct margins. The appearance closely resembles WT; however, several distinctive features like subcapsular fluid collection, tumor lobules separated by dark areas of necrosis or hemorrhage and calcification outlining tumor lobules suggest the diagnosis. Rhabdoid tumor has the worst prognosis of all renal tumors, and ~80% patients develop metastases to lung, liver, brain and skeleton. There can be associated primary brain lesion mainly in the posterior fossa (medulloblastoma or PNET). Survival is poor, with an 18-month survival rate of only 20%.[1,2]
Mesoblastic nephroma
For many years, it was believed that WT in neonates had a better prognosis than when it occurred in older children. In the late 60s, it was realized that most of these congenital renal tumors were not WT but a distinct clinical and pathologic entity, the mesoblastic nephroma.[12]
Mesoblastic nephroma is the most common solid renal tumor in neonates. It usually presents in the first 3 months of life, with a male preponderance, and ~90% of the cases are seen in the first year of life.[1,2] Some cases are detected at prenatal USG. There is an association with polyhydramnios, hydrops, hyper-reninemia and premature delivery. It is an unencapsulated tumor with whorled appearance on cut-section, like uterine myoma. Histopathology shows spindle-shaped mesenchymal cells and embryonal metaplasia of entrapped renal tissue. Uncommon cellular type shows increased malignant potential.[2]
On imaging, it is seen as a solid intrarenal mass typically involving the renal sinus. The mass may contain cystic, hemorrhagic and necrotic areas [Figure 14]. Local infiltration of perinephric tissues is common.[2]
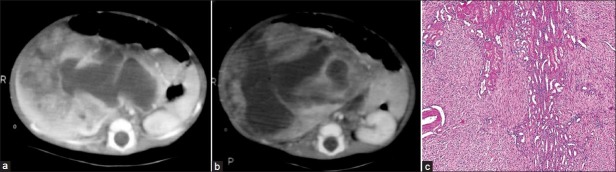
| Fig. 14 (a-b) Mesoblastic nephroma in a 2.5-month-old male – heterogeneously enhancing mass replacing right kidney with predominant cystic component. On gross pathology, the mass showed variegated areas of necrosis and cystic degeneration. Normal kidney is not seen (c) Photomicrograph – unencapsulated tumor is seen comprising predominantly of spindle cells and few entrapped tubular and glomerular components
Treatment is nephrectomy with wide surgical margin due to the infiltrative nature of the mass. Rarely, local recurrence and metastases to lung, brain or bones may be seen. Prognosis is best when diagnosed and resected before 6 months of age.[2]
Cystic partially differentiated nephroma
Cystic partially differentiated nephroma (CPDN) comes under the category of multilocular cystic renal tumors, which includes cystic nephroma (CN) and CPDN. These are generally benign lesions that cannot be differentiated by gross and radiographic appearance. However, on histopathology, septae in CN are fibrous and can have mature tubular structures and, in CPDN, the septae contain blastemal cells with or without other embryonal stromal cells.[1,2]
On sonography, CPDN is seen as a well-circumscribed encapsulated mass comprising of multiple variable-sized cysts. CECT shows enhancing septae [Figure 15a] and no excretion of contrast into loculi. When cystic spaces are small, the tumor may appear solid.[1,2]
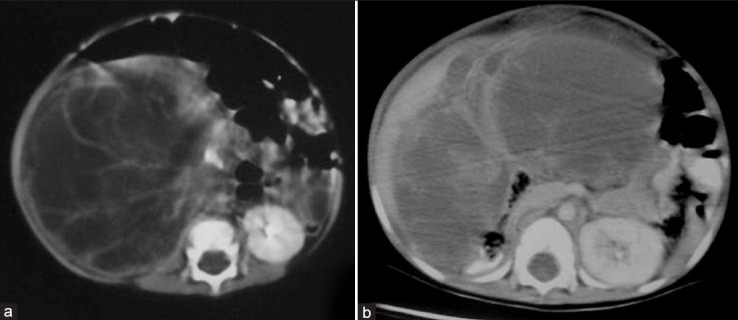
| Fig. 15 Cystic partially differentiated nephroma in a 10-month-old child with abdominal distension for 1 month – (a) contrast-enhanced computed tomography shows a multiseptate cystic mass with enhancement of septae. On surgery, the mass could not be excised completely. (b) Recurrence of cystic partially differentiated nephroma after 18 months
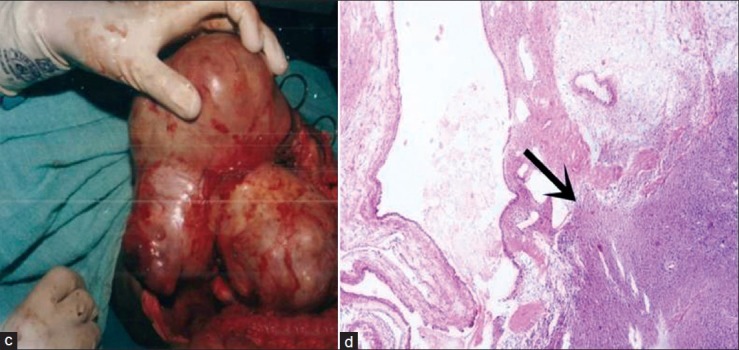
| Fig. 15 (c-d) resected specimen and photomicrograph showing cystic mass with septae showing blastemal component and abortive tubules
CPDN may demonstrate an aggressive behavior with a potential for recurrence following resection [Figure 15b]. Follow-up should be performed with non-invasive imaging methods.[1] In most cases, prognosis is excellent, with metastases being rare.[1,2]
CONCLUSION
WT is the most common malignant tumor of the kidney in children. However, in the first 3–6 months of life, mesoblastic nephroma is the most common renal neoplasm. Nephroblastomatosis, i.e. diffuse or multifocal involvement of kidney by NRs, may undergo spontaneous regression and these are more commonly associated with bilateral WT. The imaging appearance of rhabdoid tumor and clear cell sarcoma is similar to that of WT; however, clear cell sarcomas are frequently associated with skeletal metastases and rhabdoid tumor has association with brain neoplasms. Cystic nephroma and cystic partially differentiated nephroma can only be differentiated on histology as they are indistinguishable on imaging and on gross pathology. In spite of these limitations, imaging plays an important role in the management of these tumors. The extent of the tumor, locoregional and distant spread, involvement of renal vessels and the status of contralateral kidney are well demonstrated by imaging studies.
| Fig. 1 Photomicrograph showing triphasic nephroblastoma with blastemal, stromal and epithelial components
| Fig. 2(a and b) Wilms’ tumor – transverse ultrasonography shows solid mass with anechoic necrotic areas and echogenic pseudocapsule
| Fig. 3 Ultrasonography showing caval thrombus with extension into the right atrium in a case of Wilms’ tumor
| Fig. 4 Stage V Wilms’ tumor in a 2-year-old child – bilateral heterodense renal masses showing extensive necrosis and calcification. Right renal mass is extending into pelvicalyceal system. Bilateral renal vessels are normal. Computed tomography urography image shows excretion from left kidney. Pre-operative chemotherapy followed by right nephroureterectomy and left partial nephrectomy was done. It was Stage III tumor on the right side and Stage II tumor on the left side
| Fig. 5 Stage I Wilms’ tumor (WT) – non-contrast (NCCT) and contrast-enhanced CT (CECT) images show a large well-encapsulated left lower pole renal mass with hypodense areas on NCCT and heterogeneous enhancement on post-contrast image. Claw sign – characteristic of WT is also seen. No vascular invasion is evident. Mass enhances less than normal renal parenchyma. It was a completely resectable tumor with tumour-free margins
| Fig. 6 Stage III Wilms’ tumor – contrast-enhanced computed tomography (CECT) showing large heterogeneously enhancing mass with calcification, vascular encasement, periportal and retroperitoneal lymphadenopathy and pancreatic involvement. Multiplanar reconstruction and maximum intensity projection image shows the entire extent of mass extending till the diaphragm and spleen. The tumor is severely encasing the vessels and is densely adherent to diaphragm and pancreas; hence, it is non-resectable
| Fig. 8 (a–c) Stage III Wilms’ tumor in a 3-year-old child with hematuria. Heterogeneous mass with calcification, extending into the pelvicalyceal system and ureter with tumor embolus in urinary bladder. Extension to ureter is better appreciated on multiplanar reformatting (MPR) image (d). Photomicrograph (e) showing teratoid tumor with focal areas of osteoid, hyaline and smooth muscle differentiation
| Fig. 7 Stage III Wilms’ tumor – large lobulated heterogeneously enhancing right renal mass with renal vein and IVC thrombus extending into right atrium. Retroperitoneal lymphadenopathy, ascites and peritoneal involvement are seen
| Fig. 9 Chest X-ray and computed tomography scan of a case of Stage IV Wilms’ tumor – pulmonary nodules are not seen on X-ray chest
| Fig. 10 Nephroblastomatosis – progression to bilateral Wilms’ tumors. Right pleural effusion, mediastinal lymphadenopathy, bilateral renal masses, retroperitoneal lymphadenopathy and ascites are seen
| Fig. 11 Nephroblastomatosis – ultrasonography (USG) and computed tomography images of a 4-year-old female showing bilateral enlarged kidneys with lobulated contour. Nephrogenic rests are seen as hypoechoic lesions on USG and as hypodense, non-enhancing lesions on contrast-enhanced computed tomography (CECT). They are better appreciated on CECT
| Fig. 12 Nephroblastomatosis – after a 2-month interval, spontaneous regression of some lesions is evident. Renal size is decreased bilaterally and hypodense lesions (nephrogenic rests) are better appreciated
| Fig. 13 Clear cell sarcoma kidney – heterogeneously enhancing right renal mass with extensive necrosis
| Fig. 14 (a-b) Mesoblastic nephroma in a 2.5-month-old male – heterogeneously enhancing mass replacing right kidney with predominant cystic component. On gross pathology, the mass showed variegated areas of necrosis and cystic degeneration. Normal kidney is not seen (c) Photomicrograph – unencapsulated tumor is seen comprising predominantly of spindle cells and few entrapped tubular and glomerular components
| Fig. 15 Cystic partially differentiated nephroma in a 10-month-old child with abdominal distension for 1 month – (a) contrast-enhanced computed tomography shows a multiseptate cystic mass with enhancement of septae. On surgery, the mass could not be excised completely. (b) Recurrence of cystic partially differentiated nephroma after 18 months
| Fig. 15 (c-d) resected specimen and photomicrograph showing cystic mass with septae showing blastemal component and abortive tubules