 PDF
PDF  Views
Views  Share
Share


Multicentric sinus histicytosis (Rosai-Dorfman Disease): Computed tomography, magnetic resonance imaging findings
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2011; 32(03): 174-176
DOI: DOI: 10.4103/0971-5851.92826
Abstract
Sinus histicytosis (Rosai-Dorfman disease) is a rare, benign granulomatous disease that typically presents with massive cervical lymphadenopathy. Extranodal involvement along with concomitant nodal disease occurs in about 43% of cases. We present a unique case of a 30-year-old male with cervical, retroperitoneal lymphadenopathy, associated with renal and pituitary sellar masses.
Keywords
Computed tomography - magnetic resonance imaging - Rosai-Dorfman disease - sinus histicytosisPublication History
Article published online:
06 August 2021
© 2011. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Sinus histicytosis (Rosai–Dorfman disease) is a rare, benign granulomatous disease that typically presents with massive cervical lymphadenopathy. Extranodal involvement along with concomitant nodal disease occurs in about 43% of cases. We present a unique case of a 30-year-old male with cervical, retroperitoneal lymphadenopathy, associated with renal and pituitary sellar masses.
INTRODUCTION
Rosai–Dorfman disease is a benign histiocytic proliferation that can present with large lymph node masses, most often cervical.[1] The extranodal disease occurs in about 43% and it usually includes skin, soft tissues, respiratory system, genitourinary system, bones, central nervous system (CNS), orbit, thyroid and breast,[2–9] but CNS and renal involvement is very rare. The diagnosis requires histologic examination, which shows intrasinus histiocytic proliferation with cells showing emperipolesis or lymphocytophagocytosis. We present a unique case of multicentric Rosai–Dorfman disease with computed tomography (CT) and magnetic resonance imaging (MRI) findings.
CASE REPORT
A 30-year-old man presented with headache, vomiting and diminished vision for 2 years since 2005. On examination, there was multiple cervical lymphadenopathy, which were discrete, firm, mobile, nontender, along with enlargement of submandibular glands. There was no hepatosplenomegaly. Contrast-enhanced CT (CECT) scan of head, neck, chest and abdomen demonstrated an intensely enhancing mass in pituitary, cervical lymphadenopathy [Figure [Figure1a1a and andb],b], retroperitoneal lymphadenopathy and left renal mass [Figure [Figure1c1c and andd].d]. MRI brain showed the pituitary mass which was isointense on T1W and T2W images and showed intense contrast enhancement post gadolinium administration [Figure 2]. Fine needle aspiration cytology (FNAC) from cervical lymph nodes showed features of sinus histiocytosis.
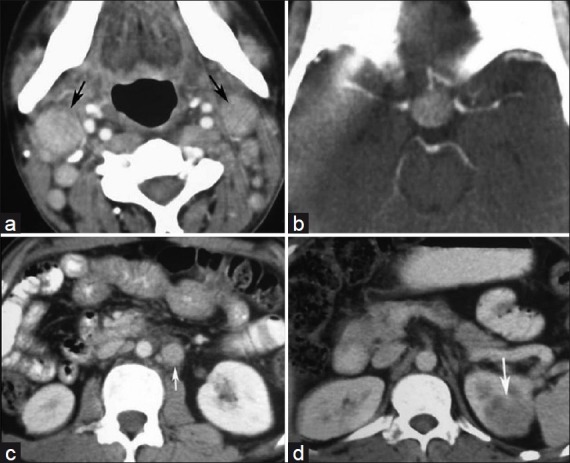
| Fig. 1 (a) Axial CECT of neck shows enhancing right cervical lymphadenopathy (black arrow); (b) Axial CECT of head shows intensely enhancing pituitary mass; (c) Axial CECT of lower abdomen showing enhancing paraaortic lymphadenopathy (white arrow); (d) Axial CECT of abdomen showing hypodense left renal mass (white arrow)
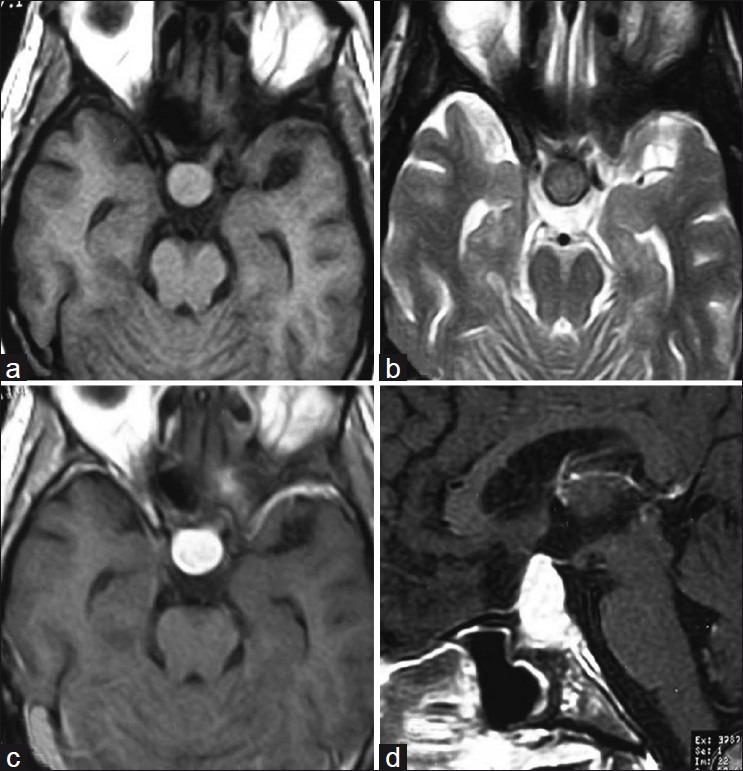
| Fig. 2 (a) Axial T1W image showing isointense pituitary mass; (b) Axial T2W showing isointense pituitary mass; (c-d) Contrast-enhanced T1W showing intensely enhancing pituitary mass in axial and sagittal sections
The patient underwent transphenoidal pituitary tumor decompression and biopsy of pituitary mass. Histopathology showed sheets of foamy histiocytes admixed with mature lymphocytes and occasional plasma cells [Figure 3]. No eosinophils or identifiable pituitary parenchyma cells were noted by light microscopy. In addition, an interesting finding noted was traversing lymphocytes and few plasma cells through cytoplasm of histiocytes (emperipolesis). On immunohistochemistry, the histiocytes were diffusely positive for CD 68 and S-100 protein and were negative for CD 1a. Only few chromogranin positive residual pituitary parenchyma cells were noted amidst the sheets of histiocytes. These features were consistent with Rosai–Dorfman disease.
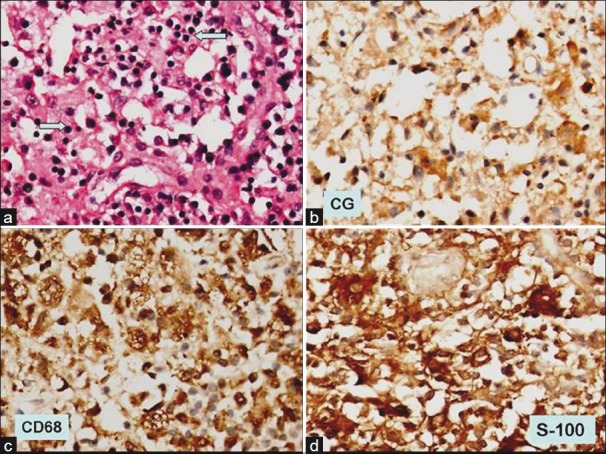
| Fig. 3 (a) Photomicrograph shows replacement of normal pituitary parenchyma by sheets of histiocytes showing emperipolesis (blue arrow) (H and E, ×200); (b) immunohistochemistry for chromogranin shows occasional immunopositive pituitary parenchyma cells, (×200); (c) the histiocytes are strongly positive for CD 68 immunohistochemistry, (×200); (d) as well the histiocytes are positive for S-100 protein (×200)
DISCUSSION
Sinus histiocytosis with massive lymphadenopathy (SHML) is called as Rosai–Dorfman disease, a rare histiocytic proliferative disorder of unknown etiology. It occurs in children and adults with mean age of incidence 20.6 years.[2] Histological features currently define it. Typically, it is characterized by bilateral cervical lymphadenopathy with fever, leukocytosis, increased erythrocyte sedimentation rate and hypergammaglobulinemia.[2] It is a multifocal and multisystem condition in 43% of cases. The extranodal involvement usually occurs with lymphadenopathy. It affects more commonly males (1.4:1).[2,3] Patients with soft tissue involvement tend to be older than patients who present with other extranodal sites.[3] In the absence of lymphadenopathy, diagnosis of SHML is difficult both clinically and pathologically because of the lack of the most typical manifestation of the disease. The histological appearance is characteristic for this disease and is essentially similar regardless of the site of occurrence.[4] The characteristic pathologic feature of this disease is proliferation of distinctive histiocytic cells that demonstrate emperipolesis in the background of a mixed inflammatory infiltrate consisting of moderately abundant plasma cells and lymphocytes. These histiocytes have a normal activated phenotype. Association with immunological abnormalities or autoimmune events, most often autoimmune cytopenia, is possible. This association is a poor prognostic factor. Although emperipolesis is not unique to this condition, in the appropriate clinical and pathologic setting, the presence of this phenomenon in the histiocytes that express S-100 protein is considered diagnostic of Rosai–Dorfman disease.
The etiology, pathogenesis and natural history of Rosai–Dorfman disease are unknown. An immune-mediated origin is proposed.[10] Numerous reports have identified human herpes virus 6 (HHV-6) in visceral and cutaneous lesions.[11] However, HHV-6 has frequently been found in many reactive and infectious disorders of lymphoid tissue, and its presence in Rosai–Dorfman disease is non-specific. With Rosai–Dorfman being a multiorgan disorder, the clinical manifestations vary and so do the radiological features, depending on the site involvement.[5] The imaging features are occasionally confounding because of the differential diagnostic possibilities. The diagnosis of Rosai–Dorfman is seldom considered, largely attributable to the rarity of the disease and low index of suspicion. The differential diagnosis for sellar and suprasellar mass includes germinoma, granulomatous disease, other histiocytoses (Langerhans cell histiocytosis), and metastasis.[12] The differential diagnosis in renal mass includes renal neoplasms like renal cell carcinoma.[13] The lymphadenopathy shows typical enhancing pattern which can be differentiated from other diseases like tuberculosis and lymphoma. There is no ideal therapeutic regimen for this disease.[10] The treatment options range from surgery, radiotherapy and steroids to chemotherapy. Treatment is reserved for forms that are directly threatening, progressive, or when poor prognostic factors are present.
However, a number of questions dealing with the preoperative diagnosis, the surgical strategies, as well as the natural history, the prognosis of the disease, and the possible need for adjunctive therapy remain obscure. Additional reports are useful to better understand this rare type of disease and may lead to the establishment of a more profitable management. To our knowledge, this is a rare case of Rosai–Dorfman involving pituitary and kidney in addition to lymphadenopathy. Rosai–Dorfman disease should be considered in the differential diagnosis of granulomatous infection, pseudogranulomatous lesion and malignancy.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
| Fig. 1 (a) Axial CECT of neck shows enhancing right cervical lymphadenopathy (black arrow); (b) Axial CECT of head shows intensely enhancing pituitary mass; (c) Axial CECT of lower abdomen showing enhancing paraaortic lymphadenopathy (white arrow); (d) Axial CECT of abdomen showing hypodense left renal mass (white arrow)
| Fig. 2 (a) Axial T1W image showing isointense pituitary mass; (b) Axial T2W showing isointense pituitary mass; (c-d) Contrast-enhanced T1W showing intensely enhancing pituitary mass in axial and sagittal sections
| Fig. 3 (a) Photomicrograph shows replacement of normal pituitary parenchyma by sheets of histiocytes showing emperipolesis (blue arrow) (H and E, ×200); (b) immunohistochemistry for chromogranin shows occasional immunopositive pituitary parenchyma cells, (×200); (c) the histiocytes are strongly positive for CD 68 immunohistochemistry, (×200); (d) as well the histiocytes are positive for S-100 protein (×200)