 PDF
PDF  Views
Views  Share
Share


Primary Adrenal Leiomyosarcoma: An Extremely Rare Mesenchymal Tumor
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2019; 40(04): 559-562
DOI: DOI: 10.4103/ijmpo.ijmpo_270_19
Introduction
Leiomyosarcoma is a soft-tissue neoplasm of smooth muscle origin. Primarily, it occurs in the myometrium, retroperitoneum, or dermis of the extremities; primary leiomyosarcoma of the adrenal gland is very rare. Till date, only 30 patients have been reported. Here, we report another case of primary adrenal leiomyosarcoma and review of the clinical and pathological characteristics.
Publication History
Received: 25 December 2019
Accepted: 06 January 2020
Article published online:
03 June 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Introduction
Leiomyosarcoma is a soft-tissue neoplasm of smooth muscle origin. Primarily, it occurs in the myometrium, retroperitoneum, or dermis of the extremities; primary leiomyosarcoma of the adrenal gland is very rare. Till date, only 30 patients have been reported. Here, we report another case of primary adrenal leiomyosarcoma and review of the clinical and pathological characteristics.
Case Report
A 60-year-old female presented with a 2 years’ history of left abdominal pain radiating to the back. There were no constitutional symptoms, as well as recent weight loss. There was no history of hypertension, diabetes, and tuberculosis, and her HIV antibody was negative. Physical examination showed mild abdominal discomfort on palpation. Computed tomography was performed, which revealed a well-circumscribed heterogeneous mass measuring 7.6 cm × 7.7 cm × 6.8 cm located in the left suprarenal area, abutting distal pancreas, and kidney [Figure 1]. Neither venous thrombosis nor metastatic lesions were noted. Twenty-four hours urine collections for cortisol and catecholamines were normal, as were serum aldosterone and adrenocorticotrophic hormone levels.
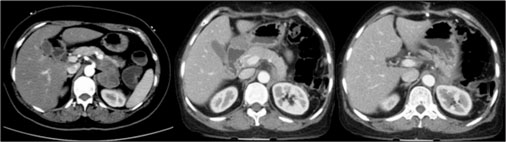
| Fig. 1 Preoperative and postoperative images of left adrenelectomy
Question 1
What are the most common causes of such a presentation?
Answer
Benign adrenocortical adenoma
Adrenocortical carcinoma
Pheochromocytoma/paraganglioma.
Other rare causes are neuroblastoma and nonprimary adrenal masses such as adrenal metastases, sarcomas, lymphomas, myelolipomas, and ganglioneuromas.
Based on the clinical diagnosis of nonfunctional adrenal tumor, a left adrenalectomy with splenectomy and distal pancreatectomy was performed.
Pathological examination: Gross pathological examination showed an encapsulated solid mass measuring roundish 9 cm × 8 cm × 7 cm. The tumor tightly abutted the stretched identifiable adrenal gland and part of the pancreas. There were areas of hemorrhage. The microscopic examination revealed a spindle cell neoplasm. There was necrosis with a mitotic rate of 5–6/10 HPF. There was a lesion abutting adjacent pancreas. Margins were free.
Morphologically adrenal gland with tumor surface interface [Figure 2a], low power spindle fascicle with pleomorphic cell [Figure 2b], On immunohistochemical studies, the tumor cells stained positive for smooth muscle actin (SMA) [Figure 2c], desmin [Figure 2d], h-Caldesmon [Figure 2e] and CD34, and negative for CD117, S100, and DOG 1. The proliferation rate of ki67 was high (40%) [Figure 2f]. Based on the histopathological and immunohistochemical findings, the diagnosis of an adrenal leiomyosarcoma was made.
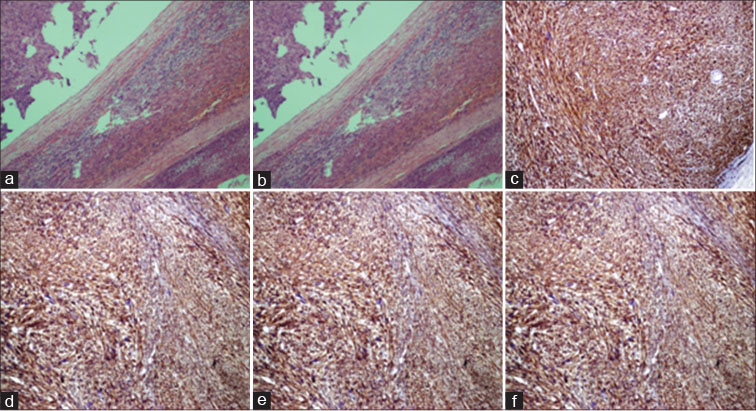
| Fig. 2 (a-f) Adrenal gland with tumor interface, low power spindle fascicle with pleomorphic cell, smooth muscle actin, Desmin, h-Caldesmin, and Ki 67, respectively
Question 2
What are the differential diagnosis of Primary Leiomyosarcoma of adrenal gland (PLAG)
Reroperitoneal or metastatic leiomyosarcoma
Primary rhabdomyosarcoma
Undifferentiated pleomorphc sarcoma.
The postoperative period was uneventful; the patient was planned for adjuvant chemotherapy with ifosfamide with Adriamycin regimen.
Question 3
How common is leiomyosarcoma of the adrenal gland?
PLAG is extremely rare and was first described by Choi and Liu in 1981.[1] Till date, only 30 cases of PLAG have been reported [Table 1].
Table 1 Summary of previously reported cases of primary adrenal leiomyosarcoma
Discussion
Almost all clinically reported cases to date are elderly patients with large tumors. It is believed to originate from the smooth muscle wall of the central adrenal vein and its branches. Most patients present with abdominal or flank pain and some also present with lower limb edema and spider angioma when the tumor invades to the inferior vena cava. Incidence of Leiomyosarcoma of adrenal gland is similar in male and female. There is equal probability of either right or left adrenal gland origin. Most of the reported cases are of conventional type and only five cases are of pleomorphic variety.
It is interesting to note that four of the patients were immunodeficient due to HIV or Epstein–Barr virus infection. It seems that primary adrenal leiomyosarcoma (PAL) is likely to occur in an immunosuppressive situation, but nothing certain is known about the pathogenic involvement of these viruses.
As PLAGs do not produce any adrenal hormonal derangement and grow rapidly, there are no applicable tumor markers or imaging characteristics available for making a preoperative diagnosis, and all the cases were diagnosed after surgery or at necropsy. Encouragingly, Goto et al. reported a case of neuron-specific enolase (NSE)-producing PAL. The level of serum NSE was markedly high preoperatively and NSE protein was massively expressed in the resected tumor. After the surgery, serum NSE levels became normal. It is suggested that serum NSE levels could be a useful hallmark for the early detection for PAL. However, Kato et al. found that immunostaining for NSE was negative in their case. Hence, we need further research to seek a suitable tumor marker.
Histopathological and immunohistochemical evaluation is indispensable not only for determining tumor type but also for differential diagnosis. Conventional leiomyosarcomas show strong immunoreactivity for smooth muscle markers such as SMA and/or muscle-specific actin in 90%–95% of cases and desmin in 70%–90% of cases. However, there is a marked variability in the expression of these markers in pleomorphic leiomyosarcomas. Oda et al. reported that 37.5% of the pleomorphic leiomyosarcomas of various sites are desmin positive, 46.4% are muscle-specific actin positive, and 50% are SMA positive. Malignant fibrous histiocytoma, malignant melanoma, malignant hemangioperistoma, angiosarcoma, liposarcoma, carcinosarcoma, rhabdomyosarcoma, adrenal invasion by a retroperitoneal leiomyosarcoma, and metastatic tumors should be considered in the differential diagnosis of adrenal leiomyosarcomas.
Radical surgery is the mainstay of therapy, but the prognosis for PLAG patients is not predictable. It is believed that in patients with invasive diseases that include venous thrombosis, adjacent organ invasion, and distant metastases, the prognosis is extremely poor. We found that in all these 28 cases, 12 patients who had no recurrence or metastasis were almost without invasive diseases and none of them underwent any adjuvant therapy such as chemotherapy or radiotherapy. Adjuvant therapy combined with surgery is often used for PLAG patients with poor prognosis. In a systematic overview study, Strander et al. showed that postoperative adjuvant radiation therapy was recommended for the treatment of locally advanced malignancy in soft-tissue sarcomas. Radiation therapy and/or chemotherapy may be helpful to shrink the tumor and destroy the remaining tumor cells.
Conclusion
Primary adrenal leiomyosarcoma is an extremely rare mesenchymal tumor. Early and complete surgical resection is the mainstay management with PFS benefit of adjuvant chemotherapy with ifosfamide and doxorubicin.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Conflict of Interest
There are no conflicts of interest.
References
- 1 Choi SH, Liu K. Leiomyosarcoma of the adrenal gland and its angiographic features: A case report. J Surg Oncol 1981; 16: 145-8
- 2 Lack EE, Graham CW, Azumi N, Bitterman P, Rusnock EJ, O’Brien W. et al. Primary leiomyosarcoma of adrenal gland. Case report with immunohistochemical and ultrastructural study. Am J Surg Pathol 1991; 15: 899-905
- 3 Zetler PJ, Filipenko JD, Bilbey JH, Schmidt N. Primary adrenal leiomyosarcoma in a man with acquired immunodeficiency syndrome (AIDS). Further evidence for an increase in smooth muscle tumors related to epstein-barr infection in AIDS. Arch Pathol Lab Med 1995; 119: 1164-7
- 4 Boman F, Gultekin H, Dickman PS. Latent epstein-barr virus infection demonstrated in low-grade leiomyosarcomas of adults with acquired immunodeficiency syndrome, but not in adjacent Kaposi’s lesion or smooth muscle tumors in immunocompetent patients. Arch Pathol Lab Med 1997; 121: 834-8
- 5 Etten B, van Ijken MG, Mooi WJ, Oudkerk M, van Geel AN. Primary leiomyosarcoma of the adrenal gland. Sarcoma 2001; 5: 95-9
- 6 Matsui Y, Fujikawa K, Oka H, Fukuzawa S, Takeuchi H. Adrenal leiomyosarcoma extending into the right atrium. Int J Urol 2002; 9: 54-6
- 7 Lujan MG, Hoang MP. Pleomorphic leiomyosarcoma of the adrenal gland. Arch Pathol Lab Med 2003; 127: e32-5
- 8 Thamboo TP, Liew LC, Raju GC. Adrenal leiomyosarcoma: A case report and literature review. Pathology 2003; 35: 47-9
- 9 Linos D, Kiriakopoulos AC, Tsakayannis DE, Theodoridou M, Chrousos G. Laparoscopic excision of bilateral primary adrenal leiomyosarcomas in a 14-year-old girl with acquired immunodeficiency syndrome (AIDS). Surgery 2004; 136: 1098-100
- 10 Kato T, Kato T, Sakamoto S, Kobayashi T, Ikeda R, Nakamura T. et al. Primary adrenal leiomyosarcoma with inferior vena cava thrombosis. Int J Clin Oncol 2004; 9: 189-92
- 11 Wong C, Von Oppell UO, Scott-Coombes D. Cold feet from adrenal leiomyosarcoma. J R Soc Med 2005; 98: 418-20
- 12 Candanedo-González FA, Vela Chávez T, Cérbulo-Vázquez A. Pleomorphic leiomyosarcoma of the adrenal gland with osteoclast- like giant cells. Endocr Pathol 2005; 16: 75-81
- 13 Lee CW, Tsang YM, Liu KL. Primary adrenal leiomyosarcoma. Abdom Imaging 2006; 31: 123-4
- 14 Mohanty SK, Balani JP, Parwani AV. Pleomorphic leiomyosarcoma of the adrenal gland: Case report and review of the literature. Urology 2007; 70: 591.e5-7
- 15 Wang TS, Ocal IT, Salem RR, Elefteriades J, Sosa JA. Leiomyosarcoma of the adrenal vein: A novel approach to surgical resection. World J Surg Oncol 2007; 5: 109
- 16 Goto J, Otsuka F, Kodera R, Miyoshi T, Kinomura M, Otani H. et al. A rare tumor in the adrenal region: Neuron-specific enolase (NSE)-producing leiomyosarcoma in an elderly hypertensive patient. Endocr J 2008; 55: 175-81
- 17 Mencoboni M, Bergaglio M, Truini M, Varaldo M. Primary adrenal leiomyosarcoma: A case report and literature review. Clin Med Oncol 2008; 2: 353-6
- 18 Van Laarhoven HW, Vinken M, Mus R, Flucke U, Oyen WJ, Van der Graaf WT. The diagnostic hurdle of an elderly male with bone pain: How 18F-FDG-PET led to diagnosis of a leiomyosarcoma of the adrenal gland. Anticancer Res 2009; 29: 469-72
- 19 Hamada S, Ito K, Tobe M, Otsuki H, Hama Y, Kato Y. et al. Bilateral adrenal leiomyosarcoma treated with multiple local therapies. Int J Clin Oncol 2009; 14: 356-60
- 20 Karaosmanoglu AD, Gee MS. Sonographic findings of an adrenal leiomyosarcoma. J Ultrasound Med 2010; 29: 1369-73
- 21 Shao IH, Lee WC, Chen TD, Chiang YJ. Leiomyosarcoma of the adrenal vein. Chang Gung Med J 2012; 35: 428-31
- 22 Kanthan R, Senger JL, Kanthan S. Three uncommon adrenal incidentalomas: A 13-year surgical pathology review. World J Surg Oncol 2012; 10: 64
- 23 Deshmukh SD, Babanagare SV, Anand M, Pande DP, Yavalkar P. Primary adrenal leiomyosarcoma: A case report with immunohistochemical study and review of literature. J Cancer Res Ther 2013; 9: 114-6
- 24 Gulpinar MT, Yildirim A, Gucluer B, Atis RG, Canakci C, Gurbuz C. et al. Primary leiomyosarcoma of the adrenal gland: A case report with immunohistochemical study and literature review. Case Rep Urol 2014; 2014: 489630
- 25 Oztürk H. Vena Cava ınvasion by adrenal leiomyosarcoma. Rare Tumors 2014; 6: 5275
- 26 Lee S, Tanawit GD, Lopez RA, Zamuco JT, Cheng BG, Siozon MV. Primary leiomyosarcoma of adrenal gland with tissue eosinophilic infiltration. Korean J Pathol 2014; 48: 423-5
- 27 Bhalla A, Sandhu F, Sieber S. Primary adrenal leiomyosarcoma: A case report and review of the literature. Conn Med 2014; 78: 403-7
- 28 Wei J, Sun A, Tao J, Wang C, Liu F. Primary adrenal leiomyosarcoma: Case report and review of the literature. Int J Surg Pathol 2014; 22: 722-6
Address for correspondence
Publication History
Received: 25 December 2019
Accepted: 06 January 2020
Article published online:
03 June 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Fig. 1 Preoperative and postoperative images of left adrenelectomy
| Fig. 2 (a-f) Adrenal gland with tumor interface, low power spindle fascicle with pleomorphic cell, smooth muscle actin, Desmin, h-Caldesmin, and Ki 67, respectively
References
- 1 Choi SH, Liu K. Leiomyosarcoma of the adrenal gland and its angiographic features: A case report. J Surg Oncol 1981; 16: 145-8
- 2 Lack EE, Graham CW, Azumi N, Bitterman P, Rusnock EJ, O’Brien W. et al. Primary leiomyosarcoma of adrenal gland. Case report with immunohistochemical and ultrastructural study. Am J Surg Pathol 1991; 15: 899-905
- 3 Zetler PJ, Filipenko JD, Bilbey JH, Schmidt N. Primary adrenal leiomyosarcoma in a man with acquired immunodeficiency syndrome (AIDS). Further evidence for an increase in smooth muscle tumors related to epstein-barr infection in AIDS. Arch Pathol Lab Med 1995; 119: 1164-7
- 4 Boman F, Gultekin H, Dickman PS. Latent epstein-barr virus infection demonstrated in low-grade leiomyosarcomas of adults with acquired immunodeficiency syndrome, but not in adjacent Kaposi’s lesion or smooth muscle tumors in immunocompetent patients. Arch Pathol Lab Med 1997; 121: 834-8
- 5 Etten B, van Ijken MG, Mooi WJ, Oudkerk M, van Geel AN. Primary leiomyosarcoma of the adrenal gland. Sarcoma 2001; 5: 95-9
- 6 Matsui Y, Fujikawa K, Oka H, Fukuzawa S, Takeuchi H. Adrenal leiomyosarcoma extending into the right atrium. Int J Urol 2002; 9: 54-6
- 7 Lujan MG, Hoang MP. Pleomorphic leiomyosarcoma of the adrenal gland. Arch Pathol Lab Med 2003; 127: e32-5
- 8 Thamboo TP, Liew LC, Raju GC. Adrenal leiomyosarcoma: A case report and literature review. Pathology 2003; 35: 47-9
- 9 Linos D, Kiriakopoulos AC, Tsakayannis DE, Theodoridou M, Chrousos G. Laparoscopic excision of bilateral primary adrenal leiomyosarcomas in a 14-year-old girl with acquired immunodeficiency syndrome (AIDS). Surgery 2004; 136: 1098-100
- 10 Kato T, Kato T, Sakamoto S, Kobayashi T, Ikeda R, Nakamura T. et al. Primary adrenal leiomyosarcoma with inferior vena cava thrombosis. Int J Clin Oncol 2004; 9: 189-92
- 11 Wong C, Von Oppell UO, Scott-Coombes D. Cold feet from adrenal leiomyosarcoma. J R Soc Med 2005; 98: 418-20
- 12 Candanedo-González FA, Vela Chávez T, Cérbulo-Vázquez A. Pleomorphic leiomyosarcoma of the adrenal gland with osteoclast- like giant cells. Endocr Pathol 2005; 16: 75-81
- 13 Lee CW, Tsang YM, Liu KL. Primary adrenal leiomyosarcoma. Abdom Imaging 2006; 31: 123-4
- 14 Mohanty SK, Balani JP, Parwani AV. Pleomorphic leiomyosarcoma of the adrenal gland: Case report and review of the literature. Urology 2007; 70: 591.e5-7
- 15 Wang TS, Ocal IT, Salem RR, Elefteriades J, Sosa JA. Leiomyosarcoma of the adrenal vein: A novel approach to surgical resection. World J Surg Oncol 2007; 5: 109
- 16 Goto J, Otsuka F, Kodera R, Miyoshi T, Kinomura M, Otani H. et al. A rare tumor in the adrenal region: Neuron-specific enolase (NSE)-producing leiomyosarcoma in an elderly hypertensive patient. Endocr J 2008; 55: 175-81
- 17 Mencoboni M, Bergaglio M, Truini M, Varaldo M. Primary adrenal leiomyosarcoma: A case report and literature review. Clin Med Oncol 2008; 2: 353-6
- 18 Van Laarhoven HW, Vinken M, Mus R, Flucke U, Oyen WJ, Van der Graaf WT. The diagnostic hurdle of an elderly male with bone pain: How 18F-FDG-PET led to diagnosis of a leiomyosarcoma of the adrenal gland. Anticancer Res 2009; 29: 469-72
- 19 Hamada S, Ito K, Tobe M, Otsuki H, Hama Y, Kato Y. et al. Bilateral adrenal leiomyosarcoma treated with multiple local therapies. Int J Clin Oncol 2009; 14: 356-60
- 20 Karaosmanoglu AD, Gee MS. Sonographic findings of an adrenal leiomyosarcoma. J Ultrasound Med 2010; 29: 1369-73
- 21 Shao IH, Lee WC, Chen TD, Chiang YJ. Leiomyosarcoma of the adrenal vein. Chang Gung Med J 2012; 35: 428-31
- 22 Kanthan R, Senger JL, Kanthan S. Three uncommon adrenal incidentalomas: A 13-year surgical pathology review. World J Surg Oncol 2012; 10: 64
- 23 Deshmukh SD, Babanagare SV, Anand M, Pande DP, Yavalkar P. Primary adrenal leiomyosarcoma: A case report with immunohistochemical study and review of literature. J Cancer Res Ther 2013; 9: 114-6
- 24 Gulpinar MT, Yildirim A, Gucluer B, Atis RG, Canakci C, Gurbuz C. et al. Primary leiomyosarcoma of the adrenal gland: A case report with immunohistochemical study and literature review. Case Rep Urol 2014; 2014: 489630
- 25 Oztürk H. Vena Cava ınvasion by adrenal leiomyosarcoma. Rare Tumors 2014; 6: 5275
- 26 Lee S, Tanawit GD, Lopez RA, Zamuco JT, Cheng BG, Siozon MV. Primary leiomyosarcoma of adrenal gland with tissue eosinophilic infiltration. Korean J Pathol 2014; 48: 423-5
- 27 Bhalla A, Sandhu F, Sieber S. Primary adrenal leiomyosarcoma: A case report and review of the literature. Conn Med 2014; 78: 403-7
- 28 Wei J, Sun A, Tao J, Wang C, Liu F. Primary adrenal leiomyosarcoma: Case report and review of the literature. Int J Surg Pathol 2014; 22: 722-6