 PDF
PDF  Views
Views  Share
Share


Primary Neuroendocrine Carcinoma of Kidney: Report of a Rare Case
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2020; 41(02): 264-265
DOI: DOI: 10.4103/ijmpo.ijmpo_114_18
Abstract
Primary neuroendocrine tumor (NET) of kidney is extremely rare, with <100 cases reported so far. The aim of the present case study is to discuss the clinical and pathological findings of renal NET and review of the available literature. We herein report the case of a 42-year-old female patient, who presented with pain abdomen and hematuria. Immunohistochemistry of her nephrectomy specimen was positive for chromogranin A and CD56 and negative for uroplakin II, P40, and CD10. KI index was 60%. Due to rarity of the tumor, there are no established guidelines available for its treatment. She was treated with combination chemotherapy of cisplatin and etoposide.
Keywords
Neuroendocrine tumor - rare - renalPublication History
Received: 14 May 2018
Accepted: 21 June 2018
Article published online:
23 May 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Primary neuroendocrine tumor (NET) of kidney is extremely rare, with <100 cases reported so far. The aim of the present case study is to discuss the clinical and pathological findings of renal NET and review of the available literature. We herein report the case of a 42-year-old female patient, who presented with pain abdomen and hematuria. Immunohistochemistry of her nephrectomy specimen was positive for chromogranin A and CD56 and negative for uroplakin II, P40, and CD10. KI index was 60%. Due to rarity of the tumor, there are no established guidelines available for its treatment. She was treated with combination chemotherapy of cisplatin and etoposide.
Keywords
Neuroendocrine tumor - rare - renalIntroduction
Neuroendocrine tumors (NETs) are usually found in gastrointestinal tract and respiratory system. Primary NET of kidney is extremely rare. Less than 100 cases have been reported in literature till date, mostly as case reports and only a few as case series.[1] [2] [3] We report a case of a 42-year-old female with immunohistochemically proven well-differentiated NET on postnephrectomy specimen with review of the related literature.
Case Report
A 42-year-old female patient presented to us with complaints of pain abdomen and episodic hematuria for 10 days. Her clinical examination revealed a lump in the left lumber region. On laboratory investigations, she was found to have anemia. Contrast-enhanced computed tomography of abdomen revealed an ill-defined enhancing lesion in upper and mid pole of the left kidney, with approximate size of 45 mm × 51 mm × 43 mm. Multiple subcentimeter pre- and para-aortic and mesenteric lymph nodes were present [Figure 1]. Left renal vein and inferior vena cava were normal. No history of flushing or diarrhea could be demonstrated. Family and personal history were noncontributory and she was a nonsmoker. She underwent left nephrectomy. The initial histopathological examination of the specimen revealed malignant neoplasm showing neuroendocrine features. Findings of immunohistochemistry study are shown in [Figure 1]. Tumor cells were found to be positive for chromogranin A and CD56 and negative for uroplakin II, P40, and CD10, with Ki-67 index 60% [Figure 2]. Based on these findings, a final diagnosis of well-differentiated NET was made. Metastatic workup was normal. She was started on chemotherapy with cisplatin and etoposide. Etoposide was given at a dose of 100 mg/m2/day for 3 days, and cisplatin was given at a dose of 45 mg/m2 on days 2 and 3 as a continuous intravenous infusion, repeated every 4 weeks.[4]
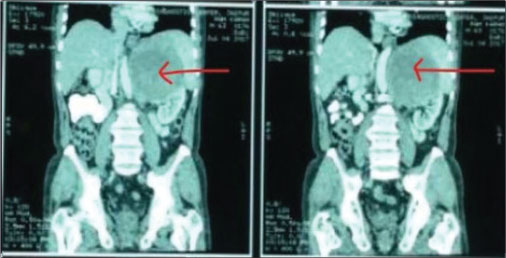
| Figure 1: Contrast-enhanced computed tomography of abdomen showing left renal mass
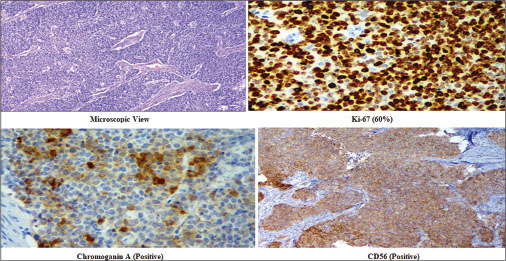
| Figure 2: Histopathological examination and immunohistochemistry of the renal lesion
Discussion
NETs are neoplasms that share a phenotype of both neuroendocrine and neural differentiation. They arise from specific neuroendocrine cells, amine precursor uptake, and decarboxylation cells. Renal NETs are unusual because neuroendocrine cells are not found in renal parenchyma. Renal NETs present a spectrum of disease: well-differentiated NET (carcinoid), well-differentiated NE carcinoma, and poorly differentiated NE carcinoma (large-cell NE carcinoma and small-cell carcinoma). Well-differentiated NETs are low-grade tumors that are slow growing; they may secrete hormones and have variable clinical course. Atypical carcinoids and poorly differentiated NETs are aggressive, present with advanced or metastatic disease, and have poor prognosis.
The pathogenesis of this tumor is uncertain because neuroendocrine cells are not found in normal adult renal parenchyma.[1] The average age of presentation is around 50 years in most of the reported cases.[5] [6] The usual presentation is flank pain, abdominal fullness, and hematuria. Right and left kidneys have been reported to be equally involved with no preference to any anatomical region. Renal NETs are usually not associated with carcinoid syndromes.[5] [6] [7] [8] About 50% of the cases present with metastasis, most commonly to lymph nodes followed by liver, lung, and bone. Grossly, they are unifocal, well demarcated, but may have focal infiltration. They are usually seen in pure form, but sometimes may be associated with teratoma. Extracapsular extension may be seen in around 50% of the cases.[5] [6] [7] [8] Microscopically, they appear as tightly packed cords, with variable stroma, nests or ribbons of uniform cells with eosinophilic, finely granular cytoplasm, and uniform nuclei. Calcification is usual, but necrosis is rare.[5] [6] [7] [8] Age more than 40 years, tumor size more than 4 cm, more than 1 mitotic figure/10 high-power field, metastasis at presentation, and extension through renal capsule have been reported as poor prognostic factors in the literature.[1]
Due to lack of randomized trials, because of the very low number of reported cases, there is no standard guideline for the treatment of renal NET. A number of options are available including debulking surgery, chemotherapy, radiotherapy, octreotide therapy, and targeted therapy.[9] Primary treatment of early-stage renal NET remains surgical excision. Large-cell NE carcinoma and small-cell carcinoma have extensive extension that might not be amenable to surgery. Residual local disease can be treated by radiotherapy and/or chemotherapy; palliation can be done for metastatic renal NETs. Combination of cisplatin and etoposide is the preferred chemotherapy for NET.[4]
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Conflict of Interest
There are no conflicts of interest.
References
- Litwinowicz R, Szpor J, Januś G, Worek M, Okoń K. Primary carcinoid tumour in horseshoe kidney. Pol J Pathol 2011; 62: 72-4
- Murali R, Kneale K, Lalak N, Delprado W. Carcinoid tumors of the urinary tract and prostate. Arch Pathol Lab Med 2006; 130: 1693-706
- Kawajiri H, Onoda N, Ohira M, Nakatani T, Wakasa K, Ishikawa T. et al. Carcinoid tumor of the kidney presenting as a large abdominal mass: Report of a case. Surg Today 2004; 34: 86-9
- Fjällskog ML, Granberg DP, Welin SL, Eriksson C, Oberg KE, Janson ET. et al. Treatment with cisplatin and etoposide in patients with neuroendocrine tumors. Cancer 2001; 92: 1101-7
- Romero FR, Rais-Bahrami S, Permpongkosol S, Fine SW, Kohanim S, Jarrett TW. et al. Primary carcinoid tumors of the kidney. J Urol 2006; 176: 2359-66
- Hansel DE, Epstein JI, Berbescu E, Fine SW, Young RH, Cheville JC. Renal carcinoid tumor: A clinicopathologic study of 21 cases. Am J Surg Pathol 2007; 31: 1539-44
- Lane BR, Jour G, Zhou M. Renal neuroendocrine tumors. Indian J Urol 2009; 25: 155-60
- Lane BR, Chery F, Jour G, Sercia L, Magi-Galluzzi C, Novick AC. et al. Renal neuroendocrine tumours: A clinicopathological study. BJU Int 2007; 100: 1030-5
- Korkmaz T, Seber S, Yavuzer D, Gumus M, Turhal NS. Primary renal carcinoid: Treatment and prognosis. Crit Rev Oncol Hematol 2013; 87: 256-64
Address for correspondence
Publication History
Received: 14 May 2018
Accepted: 21 June 2018
Article published online:
23 May 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Figure 1: Contrast-enhanced computed tomography of abdomen showing left renal mass
| Figure 2: Histopathological examination and immunohistochemistry of the renal lesion
References
- Litwinowicz R, Szpor J, Januś G, Worek M, Okoń K. Primary carcinoid tumour in horseshoe kidney. Pol J Pathol 2011; 62: 72-4
- Murali R, Kneale K, Lalak N, Delprado W. Carcinoid tumors of the urinary tract and prostate. Arch Pathol Lab Med 2006; 130: 1693-706
- Kawajiri H, Onoda N, Ohira M, Nakatani T, Wakasa K, Ishikawa T. et al. Carcinoid tumor of the kidney presenting as a large abdominal mass: Report of a case. Surg Today 2004; 34: 86-9
- Fjällskog ML, Granberg DP, Welin SL, Eriksson C, Oberg KE, Janson ET. et al. Treatment with cisplatin and etoposide in patients with neuroendocrine tumors. Cancer 2001; 92: 1101-7
- Romero FR, Rais-Bahrami S, Permpongkosol S, Fine SW, Kohanim S, Jarrett TW. et al. Primary carcinoid tumors of the kidney. J Urol 2006; 176: 2359-66
- Hansel DE, Epstein JI, Berbescu E, Fine SW, Young RH, Cheville JC. Renal carcinoid tumor: A clinicopathologic study of 21 cases. Am J Surg Pathol 2007; 31: 1539-44
- Lane BR, Jour G, Zhou M. Renal neuroendocrine tumors. Indian J Urol 2009; 25: 155-60
- Lane BR, Chery F, Jour G, Sercia L, Magi-Galluzzi C, Novick AC. et al. Renal neuroendocrine tumours: A clinicopathological study. BJU Int 2007; 100: 1030-5
- Korkmaz T, Seber S, Yavuzer D, Gumus M, Turhal NS. Primary renal carcinoid: Treatment and prognosis. Crit Rev Oncol Hematol 2013; 87: 256-64