 PDF
PDF  Views
Views  Share
Share


Rare Association of Tuberous sclerosis with Acute Lymphoblastic Leukemia: Case Report with Review of Literature
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2022; 43(04): 382-385
DOI: DOI: 10.1055/s-0042-1743126
Abstract
Acute lymphoblastic leukemia (ALL) is the most common leukemia in children in which 85%-of all cases are of B-cell ALL and approximately 15%-cases are of T-cell ALL (T-ALL). Recent revolution in next-generation sequencing has uncovered many novel somatic mutations and rearrangements in ALL cells, which have prognostic and therapeutic implications, and it has also led to recognition of germline variants in the same genes with somatic mutations commonly associated with ALL. Apart from increasing the risk of developing ALL, germline variants may influence diagnostic testing, genetic counseling, and response to antileukemic treatment. This emphasizes importance of identification of new germline variants, or association of inherited syndromes with ALL or other malignancies. Down's syndrome, Shwachman's syndrome, Fanconi anemia, Bloom's syndrome, neurofibromatosis, and ataxia telangiectasia are well-recognized conditions associated with ALL. In this communication, we report a rare association of T-ALL with tuberous sclerosis (TS). This is the first reported case, showing association of T cell leukemia and TS with confirmatory genetic work-up.
Declaration of Patient Consent
The authors certify that they have obtained all appropriate patient consent forms.
Publication History
Article published online:
28 July 2022
© 2022. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Acute lymphoblastic leukemia (ALL) is the most common leukemia in children in which 85%-of all cases are of B-cell ALL and approximately 15%-cases are of T-cell ALL (T-ALL). Recent revolution in next-generation sequencing has uncovered many novel somatic mutations and rearrangements in ALL cells, which have prognostic and therapeutic implications, and it has also led to recognition of germline variants in the same genes with somatic mutations commonly associated with ALL. Apart from increasing the risk of developing ALL, germline variants may influence diagnostic testing, genetic counseling, and response to antileukemic treatment. This emphasizes importance of identification of new germline variants, or association of inherited syndromes with ALL or other malignancies. Down's syndrome, Shwachman's syndrome, Fanconi anemia, Bloom's syndrome, neurofibromatosis, and ataxia telangiectasia are well-recognized conditions associated with ALL. In this communication, we report a rare association of T-ALL with tuberous sclerosis (TS). This is the first reported case, showing association of T cell leukemia and TS with confirmatory genetic work-up.
Keywords
acute lymphoblastic leukemia - neurocutaneous syndromes - tuberous sclerosis
Introduction
Acute lymphoblastic leukemia (ALL) is the most common pediatric malignancy with peak incidence in children between 2 and 9 years of age.[1] Recent revolution in next-generation sequencing has uncovered many novel somatic mutations and rearrangements in ALL cells, which have prognostic and therapeutic implications.[2] [3] Similarly, it has led to recognition of germline variants in the same genes (e.g., PAX5, ETV6, and IKZF1) with somatic mutations commonly associated with ALL ([Table 1]).[4] [5] Apart from increasing the risk of developing ALL, germline variants may influence diagnostic testing, genetic counseling, and response to antileukemic treatment. This emphasizes importance of identification of new germline variants, or association of inherited syndromes with ALL or other malignancies. Down's syndrome, Shwachman's syndrome, Fanconi anemia, Bloom's syndrome, neurofibromatosis, and ataxia telangiectasia are well-recognized conditions associated with ALL.[3] In this report, we describe novel association of T-cell ALL (T-ALL) with tuberous sclerosis (TS), also known as tuberous sclerosis complex (TSC). TSC is autosomal dominant neurocutaneous syndrome characterized by formation of benign hamartoma in various tissues including brain, heart, and kidneys. It is caused by mutations in tumor suppressor gene, either TSC1 or TSC2.
|
Major features |
Minor features |
|---|---|
|
Hypomelanotic macules (≥3, at least 5-mm diameter) |
“Confetti” skin lesions |
|
Angiofibromas (≥3) or fibrous cephalic plaque |
Dental enamel pits (>3) |
|
Ungual fibromas (≥2) |
Intraoral fibromas (≥2) |
|
Shagreen patch |
Retinal achromic patch |
|
Multiple retinal hamartomas |
Multiple renal cysts |
|
Cortical dysplasias[a] |
Nonrenal hamartomas |
|
Subependymal nodules |
|
|
Subependymal giant cell astrocytoma |
|
|
Cardiac rhabdomyoma |
|
|
Lymphangioleiomyomatosis[b] |
|
|
Angiomyolipomas (≥2)[b] |
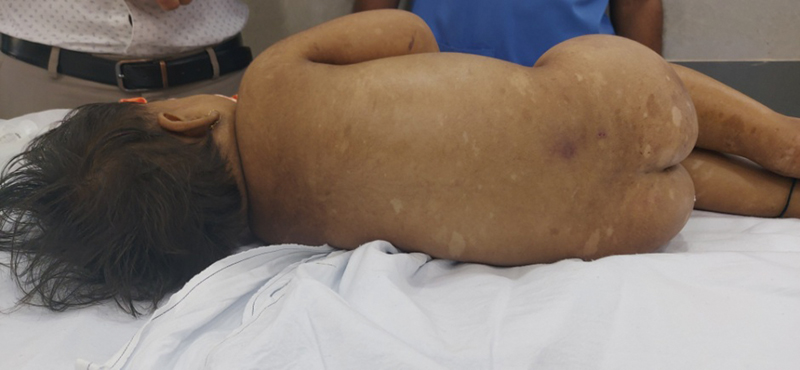
| Figure 1:Hypopigmented macules and shagreen patch on the back of patient.
On admission, laboratory investigations showed hemoglobin of 8.5 g/dL, platelet count of 9,000 cells/mm3, and white blood cells of 130,000 cells/mm3. Peripheral smear revealed a normocytic hypochromic anemia with 94% lymphoblasts. Bone marrow aspiration morphology and flowcytometry studies showing T cell markers CD1a—0.1%, CD2—93.1%, CD3 (cyto)—98.3%, CD3 (surface)—98.1%, CD4—0.4%, CD5—86.3%, CD7—84%, CD8—47.9%, TCRab—0.1%, and TCRgd—78.5% and precursor markers showing CD34—23.8%, CD117—38.5%, TdT—12.3, and CD99—15.5% confirmed the diagnosis of T-ALL. Karyotype of lymphoblast was normal and fluorescence in situ hybridization for Philadelphia (Ph) chromosome, t(12:21), and MLL rearrangement tested negative. Chest radiograph revealed a mediastinal mass. Radiological studies did not reveal presence of hamartomas anywhere in the body.
Induction chemotherapy was started as per Indian Childhood Collaborative Leukemia protocol. Child showed good response to treatment and achieved remission. At this point, whole exome sequencing was carried out on peripheral blood which revealed heterozygous exon 41 c.5226_5252delinsACT mutation in TSC2 gene on chromosome 16. The mutation is most likely germline mutation as patient was in morphological remission at the time of sampling. We could not do genetic testing on lymphoblasts and on parental samples due to financial constraints. Unfortunately, the child succumbed due to aspiration pneumonia with septic shock during further chemotherapy while leukemia was in remission.
Discussion
Neurocutaneous disorders, also described to as phacomatoses, are a group of congenital disorders with characteristic central nervous system and cutaneous manifestations presenting at different ages. These are rare diseases, with an incidence below 1:2,000 individuals in the general population. They include neurofibromatosis I and II, TSC, von Hippel–Lindau's disease, etc.[6] Owing to their genetic abnormality, they are highly susceptible to the development of tumors both benign and malignant.[7] TSC is a neurocutaneous genetic disorder with an autosomal dominant mode of inheritance and variable expressivity. Clinically cutaneous manifestations (hypopigmented macules, facial angiofibroma, shagreen patch) are most common; however, neurological (epilepsy, cognitive delay, autism) and renal (angiomyolipoma, renal cell carcinoma) manifestations are responsible for most of the morbidity and mortality associated with TSC.[8] De novo genetic mutations in TSC1 and TSC2 are known to occur in 65% of the cases. It has general prevalence of 1 in 6,000 to 10,000 newborns.[9] As far as genetic diagnostic criteria for TSC is concerned, the identification of either a TSC1 or TSC2 pathogenic mutation in DNA from normal tissue is sufficient to make a definite diagnosis of TSC. A pathogenic mutation is defined as a mutation that clearly inactivates the function of the TSC1 or TSC2 proteins (e.g., out-of-frame indel or nonsense mutation), prevents protein synthesis (e.g., large genomic deletion), or is a missense mutation whose effect on protein function has been established by functional assessment. Other TSC1 or TSC2 variants whose effect on function is less certain do not meet these criteria, and are not sufficient to make a definite diagnosis of TSC. It has been postulated that 10 to 25% of TSC patients have no mutation identified by conventional genetic testing, and a normal result does not exclude TSC, or have any effect on the use of clinical diagnostic criteria to diagnose TSC. TSC1 gene, located on chromosome 9q34, and TSC2 gene, located on chromosome 16p13.3, are tumor suppressor genes and encode for proteins called hamartin and tuberin, respectively.[10] [11] These two proteins bind each other and form a complex along with a third protein, TBC1D7 (tre2-bub2-cdc161 domain family, member 7). They play a major role in a key pathway in the cells regulating cellular homeostasis. A protein kinase called mechanistic target of rapamycin (mTOR) is involved in various cellular functions including growth, proliferation, and survival. mTOR, in turn, is controlled by Ras homolog enriched in brain (Rheb); when Rheb is activated, protein synthesis is turned on via mTOR signaling, and the cell grows. Both hamartin and tuberin are tumor suppressors, which inhibit mTOR pathway by keeping Rheb in an inactive state ([Fig. 2]).[12] Inactivation or alterations in these proteins will cause disinhibition of Rheb, leading to uncontrolled activity of mTOR pathway. Thus, somatic mutations provide “second hit” in TSC patients leading to the development of characteristic tumors such as subependymal nodules, retinal hamartomas, angiomyolipomas, rhabdomyomas, intraoral fibromas, pancreatic neuroendocrine tumors, etc.[13] Patients with TSC1 mutation are more prone for malignant tumor especially renal cell carcinoma compared with those with TSC2 mutations or no mutation.[14]
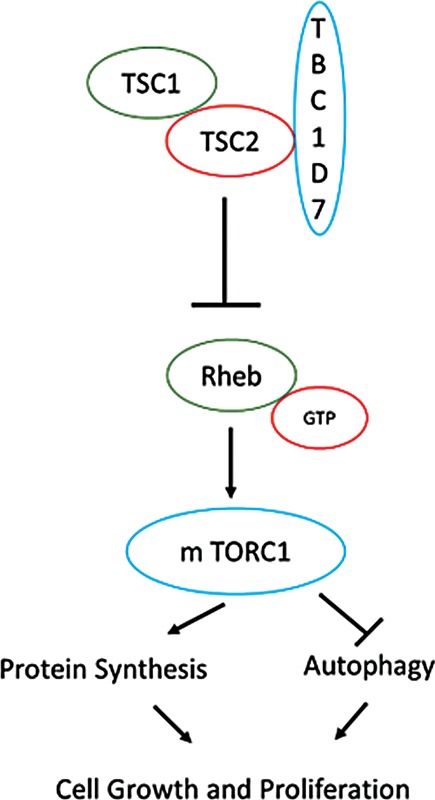
| Figure 2:Role of TSC1 and TSC2 in mechanistic target of rapamycin (mTOR) pathway and tumorigenesis.
Furthermore, mutations in several mTOR pathway component genes are known to cause specific monogenic human genetic diseases and this signaling cascade has been shown to be of relevance for Alzheimer's disease, type 2 diabetes, obesity, and hypertrophy.[15] Deregulation of these genes has also been demonstrated to be associated with sporadic bladder cancer, ovarian and gallbladder carcinoma, non-small cell carcinoma of the lung, breast cancer, pancreatic cancer, astrocytoma, xanthoastrocytoma, oral squamous cell carcinoma, and endometrial cancer.[16]
As discussed earlier, the association of TSC with tumors is well known, but hematopoietic malignancies are not commonly known to be associated with it. To our knowledge, this is the first case of association between TSC and a T-ALL. Occurrence of ALL in TSC by coincidence is less likely, as both the disorders are very rare. Alternatively, it is conceivable that there is causal relationship between TSC and T-ALL. Possibly, somatic mutation in TSC1 or TSC2 genes in hematopoietic stem cells may provide second hit, with germline mutation being first hit, to trigger leukemogenesis. Indeed, PI3K–Akt–mTOR signaling pathway is frequently upregulated in T-ALL and is associated with poor prognosis.[17] Similarly, Chiang et al have demonstrated how common β-chain-associated protein facilitates suppression of TSC2 with subsequent Rheb–mTORC1 activation in T-ALL cell line.[18] Moreover, Xu et al found that hypermethylation of TSC2 promotors led to downregulation of expression of TSC2 in acute leukemia blasts.[19] This emphasizes role of hamartin and tuberin and mTOR pathway in leukemogenesis and as possible therapeutic targets. Indeed, preclinical studies have shown promising results of activity of mTOR inhibitors in T-ALL.[20] Nonetheless, this needs further exploration with the ultimate goal of its clinical application. We could not study PI3K–Akt–mTOR signaling pathway functional studies in our patient. However, we believe that our observation of this novel and rare association of ALL is of relevance, particularly to stimulate further research.
Conclusion
One should have a high index of suspension for malignancies in cancer predisposing syndromes. We underline the rare development of hematological malignancy in TSC.
Conflict of Interest
None declared.
Declaration of Patient Consent
The authors certify that they have obtained all appropriate patient consent forms.
References
- Katz AJ, Chia VM, Schoonen WM, Kelsh MA. Acute lymphoblastic leukemia: an assessment of international incidence, survival, and disease burden. Cancer Causes Control 2015; 26 (11) 1627-1642
- Pui CH, Nichols KE, Yang JJ. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat Rev Clin Oncol 2019; 16 (04) 227-240
- Tebbi CK. Etiology of acute leukemia: a review. Cancers (Basel) 2021; 13 (09) 2256
- Morganti S, Tarantino P, Ferraro E, D’Amico P, Duso BA, Curigliano G. Next Generation Sequencing (NGS): a revolutionary technology in pharmacogenomics and personalized medicine in cancer. Translational Research and Onco-Omics Applications in the Era of Cancer Personal Genomics 2019: 9-30
- Northrup H, Krueger DA, Roberds S, Smith K, Sampson J, Korf B, Kwiatkowski DJ, Mowat D, Nellist M, Povey S, de Vries P. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatric neurology 2013; Oct 1 49 (04) 243-54
- Korf BR, Bebin EM. Neurocutaneous disorders in children. Pediatr Rev 2017; 38 (03) 119-128
- Marjanska A, Jatczak-Gaca A, Wojtkiewicz A, Wysocki M, Styczynski J. Demographical profile and spectrum of multiple malignancies in children and adults with neurocutaneous disorders. Anticancer Res 2018; 38 (09) 5453-5457
- Portocarrero LKL, Quental KN, Samorano LP, de Oliveira ZNP, Rivitti-Machado MCDM. Tuberous sclerosis complex: review based on new diagnostic criteria. An Bras Dermatol 2018; 93 (03) 323-331
- Ebrahimi-Fakhari D, Mann LL, Poryo M. et al. Incidence of tuberous sclerosis and age at first diagnosis: new data and emerging trends from a national, prospective surveillance study. Orphanet J Rare Dis 2018; 13 (01) 117
- European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993; 75 (07) 1305-1315
- van Slegtenhorst M, de Hoogt R, Hermans C. et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997; 277 (5327): 805-808
- Lam HC, Nijmeh J, Henske EP. New developments in the genetics and pathogenesis of tumours in tuberous sclerosis complex. J Pathol 2017; 241 (02) 219-225
- Borkowska J, Schwartz RA, Kotulska K, Jozwiak S. Tuberous sclerosis complex: tumors and tumorigenesis. Int J Dermatol 2011; 50 (01) 13-20
- Peron A, Vignoli A, La Briola F. et al. Do patients with tuberous sclerosis complex have an increased risk for malignancies?. Am J Med Genet A 2016; 170 (06) 1538-1544
- Rosner M, Hanneder M, Siegel N, Valli A, Fuchs C, Hengstschläger M. The mTOR pathway and its role in human genetic diseases. Mutat Res 2008; 659 (03) 284-292
- Kim LC, Cook RS, Chen J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017; 36 (16) 2191-2201
- Lonetti A, Cappellini A, Bertaina A. et al. Improving nelarabine efficacy in T cell acute lymphoblastic leukemia by targeting aberrant PI3K/AKT/mTOR signaling pathway. J Hematol Oncol 2016; 9 (01) 114
- Chiang YJ, Liao WT, Ho KC. et al. CBAP modulates Akt-dependent TSC2 phosphorylation to promote Rheb-mTORC1 signaling and growth of T-cell acute lymphoblastic leukemia. Oncogene 2019; 38 (09) 1432-1447
- Xu Z, Wang M, Wang L. et al. Aberrant expression of TSC2 gene in the newly diagnosed acute leukemia. Leuk Res 2009; 33 (07) 891-897
- Evangelisti C, Chiarini F, McCubrey JA, Martelli AM. Therapeutic targeting of mTOR in T-cell acute lymphoblastic leukemia: an update. Int J Mol Sci 2018; 19 (07) 1878
Address for correspondence
Publication History
Article published online:
28 July 2022
© 2022. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Figure 1:Hypopigmented macules and shagreen patch on the back of patient.
| Figure 2:Role of TSC1 and TSC2 in mechanistic target of rapamycin (mTOR) pathway and tumorigenesis.
References
- Katz AJ, Chia VM, Schoonen WM, Kelsh MA. Acute lymphoblastic leukemia: an assessment of international incidence, survival, and disease burden. Cancer Causes Control 2015; 26 (11) 1627-1642
- Pui CH, Nichols KE, Yang JJ. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat Rev Clin Oncol 2019; 16 (04) 227-240
- Tebbi CK. Etiology of acute leukemia: a review. Cancers (Basel) 2021; 13 (09) 2256
- Morganti S, Tarantino P, Ferraro E, D’Amico P, Duso BA, Curigliano G. Next Generation Sequencing (NGS): a revolutionary technology in pharmacogenomics and personalized medicine in cancer. Translational Research and Onco-Omics Applications in the Era of Cancer Personal Genomics 2019: 9-30
- Northrup H, Krueger DA, Roberds S, Smith K, Sampson J, Korf B, Kwiatkowski DJ, Mowat D, Nellist M, Povey S, de Vries P. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatric neurology 2013; Oct 1 49 (04) 243-54
- Korf BR, Bebin EM. Neurocutaneous disorders in children. Pediatr Rev 2017; 38 (03) 119-128
- Marjanska A, Jatczak-Gaca A, Wojtkiewicz A, Wysocki M, Styczynski J. Demographical profile and spectrum of multiple malignancies in children and adults with neurocutaneous disorders. Anticancer Res 2018; 38 (09) 5453-5457
- Portocarrero LKL, Quental KN, Samorano LP, de Oliveira ZNP, Rivitti-Machado MCDM. Tuberous sclerosis complex: review based on new diagnostic criteria. An Bras Dermatol 2018; 93 (03) 323-331
- Ebrahimi-Fakhari D, Mann LL, Poryo M. et al. Incidence of tuberous sclerosis and age at first diagnosis: new data and emerging trends from a national, prospective surveillance study. Orphanet J Rare Dis 2018; 13 (01) 117
- European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993; 75 (07) 1305-1315
- van Slegtenhorst M, de Hoogt R, Hermans C. et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997; 277 (5327): 805-808
- Lam HC, Nijmeh J, Henske EP. New developments in the genetics and pathogenesis of tumours in tuberous sclerosis complex. J Pathol 2017; 241 (02) 219-225
- Borkowska J, Schwartz RA, Kotulska K, Jozwiak S. Tuberous sclerosis complex: tumors and tumorigenesis. Int J Dermatol 2011; 50 (01) 13-20
- Peron A, Vignoli A, La Briola F. et al. Do patients with tuberous sclerosis complex have an increased risk for malignancies?. Am J Med Genet A 2016; 170 (06) 1538-1544
- Rosner M, Hanneder M, Siegel N, Valli A, Fuchs C, Hengstschläger M. The mTOR pathway and its role in human genetic diseases. Mutat Res 2008; 659 (03) 284-292
- Kim LC, Cook RS, Chen J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017; 36 (16) 2191-2201
- Lonetti A, Cappellini A, Bertaina A. et al. Improving nelarabine efficacy in T cell acute lymphoblastic leukemia by targeting aberrant PI3K/AKT/mTOR signaling pathway. J Hematol Oncol 2016; 9 (01) 114
- Chiang YJ, Liao WT, Ho KC. et al. CBAP modulates Akt-dependent TSC2 phosphorylation to promote Rheb-mTORC1 signaling and growth of T-cell acute lymphoblastic leukemia. Oncogene 2019; 38 (09) 1432-1447
- Xu Z, Wang M, Wang L. et al. Aberrant expression of TSC2 gene in the newly diagnosed acute leukemia. Leuk Res 2009; 33 (07) 891-897
- Evangelisti C, Chiarini F, McCubrey JA, Martelli AM. Therapeutic targeting of mTOR in T-cell acute lymphoblastic leukemia: an update. Int J Mol Sci 2018; 19 (07) 1878