 PDF
PDF  Views
Views  Share
Share


Revised Myeloproliferative Neoplasms Working Group Consensus Recommendations for Diagnosis and Management of Primary Myelofibrosis, Polycythemia Vera, and Essential Thrombocythemia
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2018; 39(04): 503-515
DOI: DOI: 10.4103/ijmpo.ijmpo_88_17
Abstract
Myeloproliferative neoplasms (MPNs) are clonal disorders, derived from abnormal hematopoietic stem cells and result in an excessive production of blood cells. This MPN group of conditions encompasses different diseases with overlapping clinical and biologic similarities. The majority of the conventional therapies of MPN are palliative in nature. However, with the discovery of Janus Kinase 2 (JAK2) mutation and development of targeted JAK1/2 inhibition therapy, the therapeutic options in treatment landscape have changed dramatically. This article presents the revised Indian MPNs Working Group consensus recommendations. It highlights and brings into attention about the recent findings that have defined the state of the art of the diagnosis and therapy in the MPN area, including identification of the new driver and prognostic mutations, treatment goals in the management of myelofibrosis and polycythemia vera (PV), role of the recently approved, targeted tyrosine kinase inhibitor ruxolitinib in PV, and special issues such MPN consideration in patients with splenic vein thrombosis and the management of the disease in pregnancy.
Keywords
Essential thrombocythemia Janus kinase - myelofibrosis - myeloproliferative neoplasm - polycythemia veraPublication History
Article published online:
17 June 2021
© 2018. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Myeloproliferative neoplasms (MPNs) are clonal disorders, derived from abnormal hematopoietic stem cells and result in an excessive production of blood cells. This MPN group of conditions encompasses different diseases with overlapping clinical and biologic similarities. The majority of the conventional therapies of MPN are palliative in nature. However, with the discovery of Janus Kinase 2 (JAK2) mutation and development of targeted JAK1/2 inhibition therapy, the therapeutic options in treatment landscape have changed dramatically. This article presents the revised Indian MPNs Working Group consensus recommendations. It highlights and brings into attention about the recent findings that have defined the state of the art of the diagnosis and therapy in the MPN area, including identification of the new driver and prognostic mutations, treatment goals in the management of myelofibrosis and polycythemia vera (PV), role of the recently approved, targeted tyrosine kinase inhibitor ruxolitinib in PV, and special issues such MPN consideration in patients with splenic vein thrombosis and the management of the disease in pregnancy.
Keywords
Essential thrombocythemia Janus kinase - myelofibrosis - myeloproliferative neoplasm - polycythemia veraIntroduction
Myeloproliferative neoplasms (MPNs) are a group of clonal disorders derived from multipotent hematopoietic myeloid progenitors.[1] The World Health Organization (WHO) classification of MPNs was updated in 2016 [Table 1] and has recognized polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis (MF) as the classical Philadelphia chromosome (Ph)-negative or BCR-ABL1- negative MPNs.[2] [3] Secondary MF can result from progression of PV and ET, with a 10-year risk of post-ET MF and post-PV MF being <4 href="https://www.thieme-connect.com/products/ejournals/html/10.4103/ijmpo.ijmpo_88_17#JR_4" xss=removed>4] All the 3 types of BCR-ABL1- negative MPNs pose a higher risk of transformation to acute myeloid leukemia (AML).[5] MPNs account for the large majority of myeloid malignancies[6] with incidence rates ranging from 0.1 to 3.1 cases/100,000/year in the United States and the European Union.[6] [7] [8] Among the 3 MPN subtypes, PV is the most common (0.4–2.8/100,000/year) compared to ET (0.38–1.7/100,000/year) and MF (0.1–1/100,000/year).[6] [7] [8] Epidemiological data on the incidence and prevalence of MPNs in the developing countries, including India, are sparse.
|
Criteria |
Description |
|---|---|
|
PMF – Primary myelofibrosis; WHO – World Health Organization; BM – Bone marrow; CML – Chronic myeloid leukemia; PV – Polycythemia vera; ET – Essential thrombocythemia; Hb – Hemoglobin; MDSs – Myelodysplastic syndromes; JAK2 – Janus kinase 2; CALR – Calreticulin; LDH – Lactate dehydrogenase; RCM – Red cell mass |
|
|
WHO pre-PMF criteria Major criteria |
Megakaryocytic proliferation and atypia, without reticulin fibrosis greater than Grade 1, accompanied by increased age-adjusted BM cellularity, granulocytic proliferation, and often decreased erythropoiesis Not meeting the WHO criteria for BCR-ABL1 positive CML, PV, ET, MDSs, or other myeloid neoplasms Presence of JAK2, CALR, or MPL mutation or in the absence of these mutations, presence of another clonal marker (ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1), or absence of minor reactive BM reticulin fibrosis |
|
Minor criteria |
Presence of at least 1 of the following, confirmed in 2 consecutive determinations |
|
Anemia not attributed to a comorbid condition |
|
|
Leukocytosis >11x109/L |
|
|
Palpable splenomegaly |
|
|
LDH increased to above upper normal limit of institutional reference range |
|
|
Diagnosis |
Diagnosis of pre-PMF requires meeting all 3 major criteria, and at least 1 minor criterion |
|
WHO overt PMF criteria Major criteria |
Presence of megakaryocytic proliferation and atypia, accompanied by either reticulin and/or collagen fibrosis Grades 2 or 3 |
|
Not meeting WHO criteria for ET, PV, BCR-ABL11 CML, MDSs, or other myeloid neoplasms |
|
|
Presence of JAK2, CALR, or MPL mutation or in the absence of these mutations, presence of another clonal marker, or absence of reactive myelofibrosis |
|
|
Minor criteria |
Presence of at least 1 of the following, confirmed in 2 consecutive determinations |
|
Anemia not attributed to a comorbid condition |
|
|
Leukocytosis ≥11×109/L |
|
|
Palpable splenomegaly |
|
|
LDH increased to above upper normal limit of institutional reference range |
|
|
Leukoerythroblastosis |
|
|
Diagnosis |
Diagnosis of overt PMF requires meeting all 3 major criteria, and at least 1 minor criterion |
|
WHO PV criteria Major criteria |
Hb |
|
>16.5 g/dL in men |
|
|
>16.Q g/dL in women or |
|
|
Hematocrit |
|
|
>49% in men |
|
|
>48% in women or |
|
|
Increased RCM |
|
|
BM biopsy showing hypercellularity for age with trilineage growth (panmyelosis) including prominent erythroid, granulocytic, and megakaryocytic proliferation with pleomorphic, mature megakaryocytes (differences in size) |
|
|
Presence of JAK2V617F or JAK2 exon 12 mutation |
|
|
Minor criteria |
Subnormal serum erythropoietin level |
|
Diagnosis |
Diagnosis of PV requires meeting either all 3 major criteria, or the first 2 major criteria and the minor criterion |
|
WHO ET criteria Major criteria |
Platelet count ≥45Q×109/L |
|
BM biopsy showing proliferation mainly of the megakaryocyte lineage with increased numbers of enlarged, mature megakaryocytes with hyperlobulated nuclei. No significant increase or left shift in neutrophil granulopoiesis or erythropoiesis and very rarely minor (Grade 1) increase in reticulin fibers |
|
|
Not meeting WHO criteria for BCR-ABL1 CML, PV, PMF, MDSs, or other myeloid neoplasms |
|
|
Presence of JAK2, CALR, or MPL mutation |
|
|
Minor criteria |
Presence of a clonal marker or absence of evidence for reactive thrombocytosis |
|
Diagnosis |
Diagnosis of ET requires meeting all 4 major criteria or the first 3 major criteria and the minor criterion |
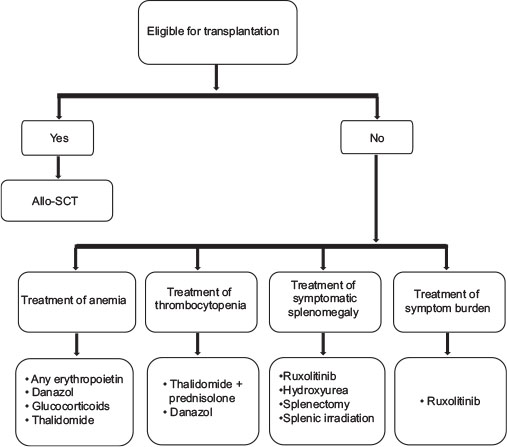
| Figure 1:Treatment algorithm for primary myelofibrosis
Risk stratification and assessing prognosis
The International Prognostic Scoring System (IPSS) is the most commonly used prognostic model for determining the survival risk category at the time of diagnosis. Based on the IPSS, patients with PMF are diagnosed as having low-, intermediate 1-, intermediate 2-, or high-risk disease. IPSS uses the following 5 risk factors to predict prognosis and assign a patient to a risk group: (1) age >65 years; (2) hemoglobin <10>2.5 × 109/L; (4) circulating blood blasts >1%; and (5) the presence of constitutional symptoms.[21] In addition, dynamic prognostic models (dynamic IPSS [DIPSS] and DIPSS Plus) are available based on the acquisition of additional risk factors during the disease course that may substantially modify patients’ outcomes.[22] [23] The DIPSS uses the same 5 risk factors and has been validated to predict prognosis at any time during the disease course. The DIPSS was recently modified (DIPSS Plus) with the incorporation of the following 3 additional risk factors: (1) red blood cell transfusion need; (2) platelet count <100 href="https://www.thieme-connect.com/products/ejournals/html/10.4103/ijmpo.ijmpo_88_17#JR_22" xss=removed>22]
Stem cell transplantation
Multiple studies have confirmed that allogeneic stem cell transplant is the only curative treatment in PMF. As this is a disease of elderly patients, only a small minority of patients are candidates for SCT. It is generally considered only for patients with intermediate-2 or high-risk disease (patients with an expected survival of <5 href="https://www.thieme-connect.com/products/ejournals/html/10.4103/ijmpo.ijmpo_88_17#JR_21" xss=removed>21] [22] [23] A transplant-eligible patient is defined as one deemed fit enough to undergo the procedure with manageable comorbidities and having an HLA-matched sibling or unrelated donor available. Patients above 45 years have a very poor survival on myeloablative conditioning.[24]
Recommendation
There are no randomized controlled trials comparing allo-HSCT to any alternative/supportive therapy. Allogeneic stem cell transplant with myeloablative or reduced intensity conditioning is indicated in young (<40 href="https://www.thieme-connect.com/products/ejournals/html/10.4103/ijmpo.ijmpo_88_17#JR_19" xss=removed>19]
Management of anemia
Erythropoietin
The use of recombinant human erythropoietin (rEPO) appears to be limited principally to a subgroup of patients with inappropriately low endogenous EPO levels (<200 href="https://www.thieme-connect.com/products/ejournals/html/10.4103/ijmpo.ijmpo_88_17#JR_19" xss=removed>19] [25] EPO levels should be measured before starting therapy with recombinant human EPO. The targeted hemoglobin level is 10 g/dL in patients with MF. EPO should be initiated at a dose of 10,000 U once weekly, and response should be monitored for 2 weeks, and if no response is observed, the dose can be escalated up to 40,000 IU once weekly. Treatment should be discontinued after 3–4 months if no response occurs. Darbepoetin has greater activity and a longer half-life than EPO, which allows for less frequent administration. Darbepoetin-α, administered bimonthly, is equally effective, and the recommended dose is in the range of 150–300 μg/fortnight. Recombinant human EPO should be stopped or dose reduced when hemoglobin is above 12 g/dL to avoid the risk of thrombosis.[19] In a recent analysis, the combination of EPO with ruxolitinib was evaluated. In this study, clinical benefits were observed in patients treated with ruxolitinib. Moreover, recently a sub-analysis of pooled data of COMFORT trials has shown that the postbaseline anemia induced by ruxolitinib in the treatment of MF has no prognostic impact on the overall survival (OS) of the patients, and the anemia due to ruxolitinib is manageable with conventional treatments than the disease-related baseline anemia, which is an established poor prognostic factor.[26]
Recommendation
EPO is recommended as the first-line therapy for the treatment of anemia in PMF only for patients with EPO levels <200 href="https://www.thieme-connect.com/products/ejournals/html/10.4103/ijmpo.ijmpo_88_17#JR_19" xss=removed>19] [25] Grade B recommendation, evidence Level III.[24]
Danazol
Danazol, a synthetic attenuated androgen, has been considered as the first-line androgen of choice in MF. Recommended starting dose is 200 mg daily, with a gradual dose escalation, depending on tolerability and patient weight (to a maximum of 600 mg daily for patients <80>80 kg). Most patients respond within the first 2–3 months. The therapy should be discontinued if no response is observed after a 4-month trial of these agents. While on danazol treatment, liver function of patients should be monitored regularly (once monthly), and the male patients should be screened for prostate cancer. A synergistic effect between human recombinant EPO and danazol treatment has been reported.[19] [27]
Recommendation
Danazol is recommended as an alternative first-line therapy in the treatment of anemia in patients with PMF associated with transfusion-dependent anemia.[19] [27] Grade B recommendation, evidence Level III.[19] [27]
Thalidomide
Low-dose thalidomide (50 mg/day preferably given at bedtime) can be used for the treatment of patients with MF with anemia and thrombocytopenia, and for the reduction of splenomegaly.[28] Low-dose thalidomide in combination with prednisolone showed significant responses for anemia (67%), thrombocytopenia (75%), and splenomegaly (33%).[29] However, thalidomide is associated with nonhematological toxicity (constipation, sedation, depression, and peripheral neuropathy) and is contraindicated in pregnancy. It is to be used with caution and with effective counseling and contraception in women of childbearing age.
Recommendation
Low-dose thalidomide (maximum 50 mg/day) in combination with prednisolone (10–20 mg/day for 2 weeks and afterward tapering to the lowest dose necessary for maintaining an adequate hemoglobin concentration) can be considered for patients not responding to EPO or danazol. Therapy should be discontinued if no response is seen after 4 months. Grade B recommendation, evidence Level III.[19]
Glucocorticoids (mostly used in combination with thalidomide)
Corticosteroids, especially prednisone, have been used for the palliation of constitutional symptoms and anemia in patients with MF. They are recommended chiefly in patients with hemolytic activity, but could be considered at a dose of 10–20 mg/day in some patients with anemia without evidence of hemolysis. In patients with hemolysis and a positive Coombs test, an initial test dose of 1 mg/kg is recommended. Grade C recommendation, evidence Level IV.[19]
The panel agreed that standard transfusion guidelines should be applied for patients eligible for transfusion to correct anemia associated with MF.
Treatment of thrombocytopenia
The panel has examined the available evidence for the treatment of thrombocytopenia associated with MF and concluded that in the context of hematology practice in India, thalidomide in combination with prednisolone should be the choice of therapy.[17] If no response is observed with this combination, danazol is recommended.
Treatment of symptomatic splenomegaly
Hydroxyurea
Hydroxyurea is one of the most commonly used agents despite the fact that the available data are limited to support its efficacy in MPN. Based on the prior experience of the panel, the members were in agreement that they have seen only modest improvement in symptoms with hydroxyurea.[30] [31] [32]
Recommendation
Hydroxyurea (0.5–2 g/d) is recommended as cytoreductive therapy (CYTO) in older patients with MF who are not eligible for transplantation. Grade B recommendation, evidence Level III.[19]
Janus kinase 2 inhibitors
JAK inhibitors are the only drugs that have been evaluated in the randomized clinical trials. A number of JAK inhibitors are at various stages of clinical development, and a consistent pattern of response in splenomegaly and disease-related symptoms is emerging.
In India, ruxolitinib is approved for the treatment of patients with MF, including primary MF, post-PV MF or post-ET MF.[33] Ruxolitinib monotherapy has been evaluated in intermediate-2-or high-risk IPSS patients with MF in phase-III COMFORT studies which have completed 5 years long-term follow-up. In COMFORT-II, 97.1%-of patients who remained on treatment had improvements from baseline in spleen volume, and 53.4%-achieved ≤35%-reductions. Spleen reductions of ⩾35%-among ruxolitinib randomized patients were sustained with long-term therapy (median duration, 3.2 years); the probability of maintaining a spleen response was 0.48 (95%-confidence interval [CI],0.35–0.60) at 5.0 years for patients initially randomized to ruxolitinib. In addition, long-term findings from COMFORT studies support the disease-modifying effect of ruxolitinib. Among patients randomized to ruxolitinib in COMFORT-II, 23 (15.8%) had improved fibrosis (any grade), 47 (32.2%) had stable fibrosis at their last assessment (median treatment duration, 2.2 years in the ruxolitinib arm). Patients randomized to ruxolitinib had longer OS compared than those randomized to best available therapy (BAT), with a 33%-reduction in risk of death with ruxolitinib treatment (HR, 0.67 (95%-CI, 0.44–1.02); P = 0.06).[34] Similar results were also found in COMFORT-I, for all ruxolitinib treated pts (n = 57), 33% (G[–1], n = 11; G [–2], n = 7; G [–3], n = 1) had an improvement, 49%-had no change or stabilization, and 18%-had a worsening in BM fibrosis from BL to the last grade.[35] A pooled OS analysis of 5-year data from the COMFORT-I and COMFORT-II shows that the risk of death was reduced by 30%-among patients randomized to ruxolitinib compared with control patients (median OS: ruxolitinib, 5.3 years; control, 3.8 years; hazard ratio [HR; ruxolitinib vs. control], 0.70; 95%-CI, 0.54 − 0.91; P = 0.0065;) as per intent-to-treat analysis and it was more pronounced after censoring patients at the time of crossover, demonstrated that ruxolitinib prolonged survival compared with control (median OS: ruxolitinib, 5.3 years; control, 2.4 years; HR [ruxolitinib vs. censored at crossover], 0.53; 95%-CI, 0.36 − 0.78; P = 0.0013).[36] The reasons for the survival advantage observed with ruxolitinib may be due to the number of benefits of treatment in terms of spleen size reduction, alleviation of cytokine-driven symptoms and inflammation, improvement of overall nutritional status, and reduced fibrosis in some patients.[34] Ruxolitinib is the current treatment of choice for constitutional prognostic symptoms of the disease (weight loss, fever, and night sweats), spleen-related symptoms (such as abdominal discomfort and pain under left ribs), and disease-related symptoms (such as itching, fatigue), as no other therapy has been shown to significantly improve symptom burden and quality of life in MF.[20] Based on these benefits including reduction of symptom burden, NCCN guidelines in the management of MPNs has recommended the use of ruxolitinib in low risk/int-1/int-2 and high-risk patients with symptoms.[37] A common misconception is that JAK inhibition is effective primarily in patients who have the JAK2 V617F mutation. However, ruxolitinib demonstrated the comparable efficacy in patients with or without the V617F mutation. Since JAK2 is involved in thrombopoietin and EPO signaling, dose-dependent thrombocytopenia and anemia should be anticipated and managed appropriately in patients with MF undergoing treatment with JAK2 inhibitors such as ruxolitinib.[34] [35] Recent updates from COMFORT studies have shown that the postbaseline anemia induced by ruxolitinib in the treatment of MF does not have an impact on the OS of the patients, and it can be managed with conventional treatments than the disease-related baseline anemia, which is an established poor prognostic factor.[26] Starting dose of ruxolitinib should be personalized according to baseline platelet counts [Table 2] and there is no separate guidance for elderly patients since both groups showed comparable clinical responses and toxicity rates similar to younger individuals, however, comorbidities have to be kept in mind while treating elderly patients with MF.[38]
|
Recommended starting doses based on platelet count |
|||||
|---|---|---|---|---|---|
|
Platelet count |
Starting dose |
||||
|
Bid – Twice daily |
|||||
|
>200×109/L |
20 mg orally bid |
||||
|
100-200×109/L |
15 mg orally bid |
||||
|
50-<100>9/L |
5 mg orally bid |
||||
|
Dose modifications (with starting platelet count of at least 100×109/L) |
|||||
|
Current platelet count |
Dose at time of decline in platelet count (new dose to be used) |
Maximum dose based on platelet count after prior treatment interruption or dose reduction |
|||
|
20 mg bid |
15 mg bid |
10 mg bid |
5 mg bid |
||
|
≥125×109/L |
No change |
No change |
No change |
No change |
20 mg bid |
|
100-<125>9/L |
15 mg bid |
No change |
No change |
No change |
15 mg bid |
|
75-<100>9/L |
10 mg bid |
10 mg bid |
No change |
No change |
10 mg bid for 2 weeks; if stable, may increase to 15 mg bid |
|
50-<75>9/L |
5 mg bid |
5 mg bid |
5 mg bid |
No change |
5 mg bid for 2 weeks; if stable, may increase to 10 mg bid |
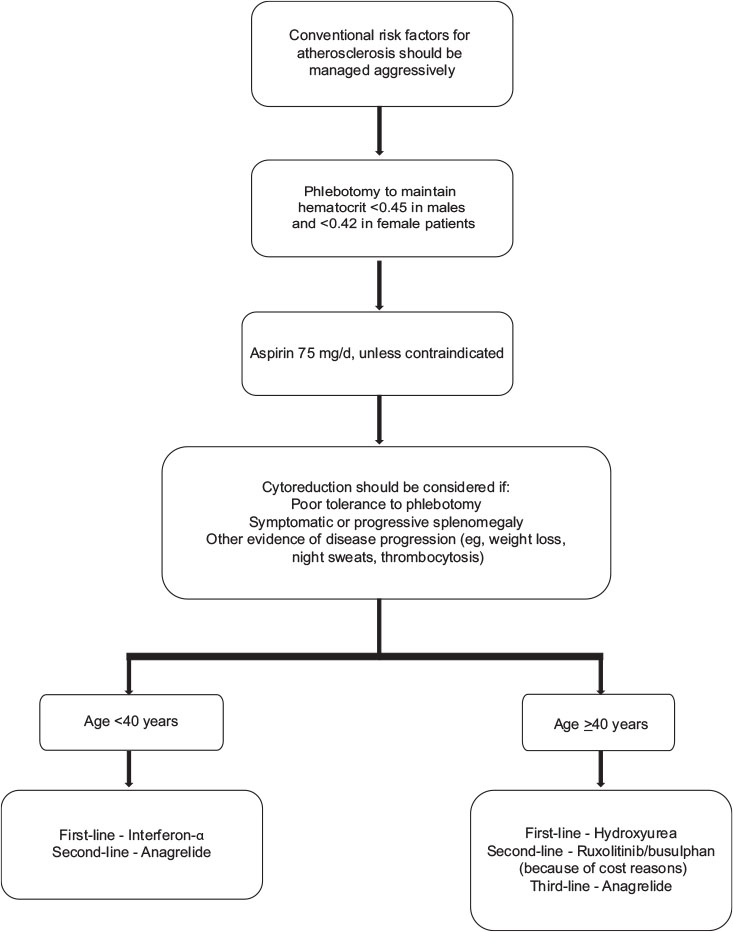
| Figure.2:Algorithm for management of essential thrombocythemia low-grade glioma subtypes
Phlebotomy
The objective of phlebotomy in PV is to improve the circulation of blood by lowering the blood viscosity. The basis of conventional therapy is to maintain hematocrit at <0 href="https://www.thieme-connect.com/products/ejournals/html/10.4103/ijmpo.ijmpo_88_17#JR_44" xss=removed>44] Phlebotomy is recommended for hematocrit levels >0.48. Grade A recommendation, evidence Level Ib.[19]
Aspirin therapy
For all patients with PV, low-dose aspirin (i.e. 100 mg daily or 81 mg in the US) is recommended. In a large randomized study, aspirin has shown superiority over placebo with a significant reduction in cardiovascular death, thrombosis, strokes, and in overall mortality.[45] Aspirin has been shown to reduce both arterial and venous thrombosis in PV.[41] Aspirin should not be given to patients with platelets more than 1500 × 109/L due to an increased risk of bleeding, instead CYTO should be initiated. In case of aspirin allergy, clopidogrel can be used. Grade A recommendation, evidence Level Ib.[19]
Choice of cytoreductive therapy in polycythemia vera
Interferon-α
Although the supporting evidence for IFN-α efficacy in PV is limited, hydroxyurea is the recommended first-line treatment for high-risk patients. Pegylated IFN-α 2a (PEG IFN-α 2a) has shown better clinical response rates and demonstrated more activity with few complete molecular remissions (as determined by JAK2 allele burden) at a better side effect profile than non-PEG versions of IFNα.[44]
Recommendation
IFN-α can be considered in younger patients (<40>40 years of age, hydroxyurea should be considered as the first-line of treatment. The panel recommends that the lower doses of IFN should be used in India because of smaller body mass index as compared to Western population and hence, the dose of PEG IFN in Indian patients may range from 40, 80, to 150 μg, weekly. Grade B recommendation, evidence Level III.[19]
Hydroxyurea
Hydroxyurea is recommended as a first-line CYTO in patients with PV >40 years or younger patients who do not tolerate IFN-α (Grade A recommendation, evidence Level Ib).[43] Hydroxyurea is useful in controlling PV-related symptoms; splenomegaly, leukocytosis, thrombocytosis, and hematocrit. However, hydroxyurea-treated patients can eventually become resistant or experience unacceptable adverse effects (hydroxyurea intolerance), including skin ulcers, a reduction in blood cells, gastrointestinal problems, oral ulcers, stomatitis, hyperkeratosis, or actinic keratosis.[46] However, hydroxyurea should be continued until patients develop intolerance or progression.[43] Increased risk of leukemic transformation has not been observed despite long-term hydroxyurea treatment. Grade A recommendation, evidence Level Ib. The panel recommended maximum tolerated dose of hydroxyurea as 1 g (this is with reference to 2 g in hydroxyurea resistance/tolerance ELN criteria).
Busulfan
Busulfan can be used as the second-line CYTO for elderly population of age >70 years. Sequential use of busulfan and hydroxyurea resulted in a significant increase in the risk of second malignancies, including leukemias.[47]
Janus kinase 2 inhibitors
Like MF landscape, several JAK inhibitors are under evaluation in clinical trials for efficacy and safety in patients with PV. Ruxolitinib is the only JAK1/2 inhibitor approved by the United States Food and administration and European Medicine Agency for the treatment of patients with PV who have had an inadequate response to or are intolerant of hydroxyurea. Findings from the pivotal RESPONSE study showed that ruxolitinib provided clinically relevant improvements compared with BAT in maintaining control of hematocrit level without phlebotomy, normalizing blood cell count, reducing spleen volume, and improving symptoms. In PV patients with inadequately controlled leukocyte count, recent evidence has shown that ruxolitinib treatment resulted in better white blood cell (WBC) control as compared to conventional therapies such as hydroxyurea (HU) and BAT.[48] Recent data from RESPONSE-II, a trial conducted in PV patients without splenomegaly, have also shown hematocrit control, complete hematologic response, and symptom control among ruxolitinib-treated patients better than BAT.[49] In addition, there were fewer thromboembolic events in the ruxolitinib arm as compared to BAT during the therapy. There is also some preliminary data on the reduction of JAK2 allele burden by ruxolitinib up to −22%-by week 80; however, the impact of this on the disease has to be studied further. The rates of all Grade and Grade 3/4 thromboembolic events per 100 patient-years of exposure were lower among patients originally randomized to ruxolitinib than BAT. The starting dose of ruxolitinib in RESPONSE was 10 mg twice daily.[48]
Recommendation
Ruxolitinib should be considered as first-choice of treatment in patients with PV who have had an inadequate response to or are intolerant of hydroxyurea. Proposed definition of resistance/intolerance to HU was defined as a dose ≤2 g/day or a maximum tolerated dose <2>400 × 109/L and WBC count >10 × 109/L; Failure to reduce splenomegaly extending >10 cm below the costal margin by >50%, as measured by palpation; Absolute neutrophil count <1 href="https://www.thieme-connect.com/products/ejournals/html/10.4103/ijmpo.ijmpo_88_17#JR_46" xss=removed>46] Grade A recommendation, evidence Level Ib.[19]
Anagrelide
Anagrelide has powerful platelet reducing activity that could be helpful in the management of patients who are intolerant or refractory to hydroxyurea, INF-α, or ruxolitinib. Grade C recommendation, evidence Level IV.[19]
Radioactive phosphorus
Due to its leukemogenic effect, radioactive phosphorus is not a recommended option for patients <75 href="https://www.thieme-connect.com/products/ejournals/html/10.4103/ijmpo.ijmpo_88_17#JR_19" xss=removed>19]
Evaluation of response and follow-up
The panel recommends that the goal of therapy should be normalization of peripheral blood counts, and the ELN criteria for PV should be used for the evaluation of response in clinical trial setting.
Essential thrombocythemia
Treatment goals
Similar to PV, the main treatment goals in ET include minimizing the risk for thrombosis and progression, normalizing peripheral blood counts, reducing constitutional symptoms, and managing special situations like pregnancy. Treatment algorithm recommended for Indian scenario is discussed in the [Figure 3].
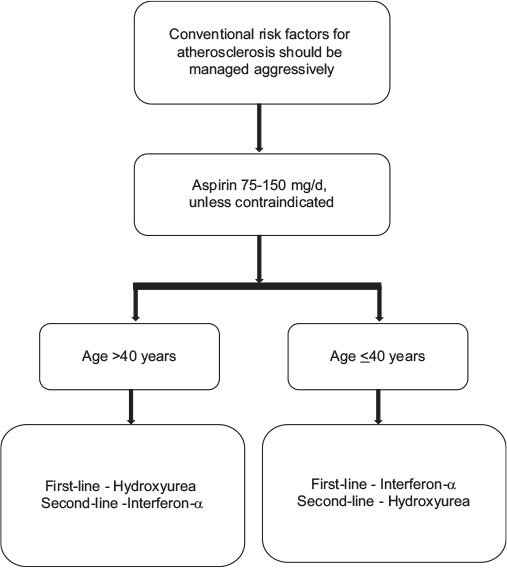
| Figure.3:Algorithm for management of polycythemia vera different low-grade glioma subtypes
Risk stratification
Risk stratification in ET is based on the assessment of risk of thrombosis, as the current therapy in ET is aimed at lowering the risk of thrombosis. True ET diagnosed, according to the 2008 WHO classification, has not been reported to affect the life expectancy of patients.[50] High-risk is defined as the presence of age above 60 years or history of previous thrombosis, or a platelet count more than 1500 × 109/L; and these patients should be treated with CYTO.[19] Low-risk is defined by the absence of these 3 factors and should not be treated with CYTO except in patients with uncontrolled cardiovascular risk factors.[19]
Clinical management
Patients with ET are at high risk for thrombosis; hence, vigorous treatment is required for managing cardiovascular risk factors. Hence, cardiovascular risk assessment is required and will dictate the overall management of the patient. It is important to emphasize that before starting therapy, patients should be evaluated for eventual progression to MF if they show symptomatic or progressive splenomegaly, other evidence of disease progression such as weight loss, night sweats, progressive leukocytosis, and/or thrombocytosis.
Stem cell transplantation is almost never performed in ET due to unfavorable risk-benefit profile. The treatment algorithm for ET is provided in [Figure 3]
Aspirin
Antiplatelet therapy with aspirin 75–150 mg/day is recommended, unless otherwise contraindicated for all the patients with ET. Aspirin should not be given to patients with platelets more than 1500 × 109/L due to an increased risk of bleeding, instead CYTO should be initiated; recommendation, evidence Level IIIB.[51]
Choice of cytoreductive therapy in essential thrombocythemia
Hydroxyurea
Hydroxyurea is the best-documented therapy in ET and is recommended as a first-line therapy in the majority of patients with high-risk ET. Hydroxyurea markedly reduces thrombotic complications compared to aspirin alone. Panel recommended that patients presenting a symptom score of <8>
Interferon-α
IFN-α treatment is well documented and safe in ET and is not considered leukemogenic or teratogenic.[51] PEG IFN has also been used; Level IB evidence to support the use of IFN.
IFN-α is the recommended first-line therapy in younger patients. It can be used in older patients if long-term use of hydroxyurea is not suitable and in patients who do not tolerate hydroxyurea (Grade B recommendation, evidence Level III). IFN-α is the treatment of choice if CYTO is indicated during pregnancy or when pregnancy is planned.
Anagrelide
This may be used as the second-line treatment in patients resistant to or intolerant of hydroxyurea. It has been found to be equivalent to hydroxyurea although some studies have reported higher risk of thrombosis.[51]
Janus kinase 2 inhibitors
Since no JAK2 inhibitor has been studied extensively in ET to date, these drugs are experimental at the present time and should not be used outside of the context of clinical trials.
Other drugs
Other drugs such as busulfan and radioactive phosphorus are not relevant in ET. Anagrelide has many complications, but may be used in some patients, if necessary, in combination with hydroxyurea to control platelet counts.
Management of complications of myeloproliferative neoplasms
Acute thrombotic events and secondary prophylaxis
In general, acute thrombotic events should be treated as in non-MPN patients. Control of hematocrit and platelet count should be optimized. In emergency situations such as acute cerebrovascular complications or severe digital ischemia, acute platelet apheresis or erythropheresis can be used in order to achieve a rapid reduction in blood counts. Since the effect is brief, CYTO preferably with hydroxyurea should be started as soon as possible in patients not on CYTO. Prevention of recurrent thrombosis should be independently achieved in patients with previous venous thrombosis by both oral anticoagulants and antiplatelet drugs. Since no prospective trials exist, it remains unclear whether it is better to give a short course of warfarin or to continue with long-term therapy for secondary prevention of venous thromboembolism.
Bleeding
The most important cause of bleeding in ET and PV is acquired von Willebrand syndrome associated with high platelet counts (>1500 × 109/L).[52] Therefore, the most important therapeutic intervention to manage acute bleeding in the patients with thrombocythemia is platelet reduction, and the recommended agent is hydroxyurea.
Platelet apheresis is indicated when extreme thrombocytosis is accompanied by an urgent need to reduce platelet counts, i.e., severe or life-threatening bleeding.[53]
Transformation to acute myeloid leukemia
The results after conventional induction chemotherapy are dismal in patients developing AML after PV, ET, or PMF, with a very short median survival.[54] If possible, it is recommended that patients undergo allo-SCT after induction chemotherapy. Hypomethylating agents can be used as bridge to transplant.[55] [56]
Splanchnic vein thrombosis and portal vein hypertension
Splanchnic vein thrombosis (SVT) includes the Budd-Chiari syndrome (BCS) and portal vein thrombosis (PVT). MPNs are the most frequent underlying prothrombotic factors in BCS and PVT. Peripheral blood cell counts often remain within a normal range because of portal hypertension and its sequelae (splenomegaly, hemodilution, and iron deficiency). In a meta-analysis, high prevalence of JAK2 V617F in SVT was reported. The strong relation between MPNs and SVT is confirmed by the high prevalence of JAK2 V617F in these patients. It is suggested that JAK2 V617F screening should be included in the routine diagnostic workup of all patients with SVT, regardless of the absence of MPN hallmarks, such as elevated peripheral blood cell counts.[57]
Special issues
Pregnancy
There is limited information in the medical literature about the management of MPNs in pregnancy.[58] The live birth rate is about 60%-due to an overall incidence of first trimester miscarriage of 31%–36% (about twice the normal rate) and an increased risk of intrauterine growth retardation, intrauterine death, and stillbirth (8%). Major maternal complications are less common and occur in approximately 8%-of patients with ET.[59]
Pregnancy is likely to be accompanied by a high risk of complications for the mother and/or fetus if any of the following factors are present: previous venous or arterial thrombosis in mother, previous hemorrhage attributed to PV/ET, previous pregnancy complications that may have been caused by PV/ET (these include significant antepartum hemorrhage or postpartum hemorrhage, severe preeclampsia, unexplained recurrent first trimester loss (≤3), intrauterine growth retardation (<5 href="https://www.thieme-connect.com/products/ejournals/html/10.4103/ijmpo.ijmpo_88_17#JR_60" xss=removed>60]
Therapeutic options include antithrombotic treatment, phlebotomy in PV, and cytoreductive agents. The optimal hematocrit in pregnancy has yet to be established, but the current recommendation is to maintain the hematocrit within the normal range appropriate for gestation. The increased plasma volume often results in a reduced hematocrit and platelet count during the second trimester. The levels rise again during the postpartum period contributing to an increased thrombotic risk during the first 6 weeks after delivery. Close monitoring of blood counts is important during this period.[56] Low-dose aspirin is safe and seems advantageous during pregnancy in ET. Starting on the day of the delivery, aspirin is substituted by a prophylactic dose of low molecular weight heparin (LMWH), which is given until 6 weeks after delivery.[54] The doses of LMWH that have been reported are dalteparin 5000 U or enoxaparin 40 mg daily.
Pediatric myeloproliferative neoplasm
The incidence of different MPN in patients aged <20>
Discussion
The diagnostic criteria and treatment landscape of MPNs are evolving at a fast pace. In this paper, the panel has appraised the recent literature and updated the existing consolidated MPN guidelines to further strengthen the understanding of fellow practitioners. The recommendations are developed solely in consensus with the experts in the field of Ph-negative classical MPNs, and a group decisionmaking process was adopted while interpreting available clinical evidence in the context of hematology practice in India. Improved diagnostic capabilities among the Indian practitioners will in turn help in identifying the true incidence and prevalence of the disease. In addition, improved diagnosis with rational treatment will help in achieving better treatment outcomes in these patients, who were previously left undiagnosed because of the insidious nature of the disease.
Conflict of Interest
There are no conflicts of interest.
References
- Levine RL, Gilliland DG. Myeloproliferative disorders. Blood 2008; 112: 2190-8
- Mascarenhas J, Mughal TI, Verstovsek S. Biology and clinical management of myeloproliferative neoplasms and development of the JAK inhibitor ruxolitinib. Curr Med Chem 2012; 19: 4399-413
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127: 2391-405
- Tefferi A. Essential thrombocythemia, polycythemia vera, and myelofibrosis: Current management and the prospect of targeted therapy. Am J Hematol 2008; 83: 491-7
- Cervantes F, Tassies D, Salgado C, Rovira M, Pereira A, Rozman C. et al. Acute transformation in nonleukemic chronic myeloproliferative disorders: Actuarial probability and main characteristics in a series of 218 patients. Acta Haematol 1991; 85: 124-7
- Visser O, Trama A, Maynadié M, Stiller C, Marcos-Gragera R, De Angelis R. et al. Incidence, survival and prevalence of myeloid malignancies in Europe. Eur J Cancer 2012; 48: 3257-66
- Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma 2014; 55: 595-600
- Moulard O, Mehta J, Fryzek J, Olivares R, Iqbal U, Mesa RA. et al. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol 2014; 92: 289-97
- Campbell PJ, Green AR. The myeloproliferative disorders. N Engl J Med 2006; 355: 2452-66
- ;Campbell PJ, Scott LM, Buck G, Wheatley K, East CL, Marsden JT. et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: A prospective study. Lancet 2005; 366: 1945-53
- Guglielmelli P, Lasho TL, Rotunno G, Score J, Mannarelli C, Pancrazzi A. et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: An international study of 797 patients. Leukemia 2014; 28: 1804-10
- Tefferi A, Guglielmelli P, Lasho TL, Rotunno G, Finke C, Mannarelli C. et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: An international study of 570 patients. Leukemia 2014; 28: 1494-500
- Agarwal MB, Malhotra H, Chakrabarti P, Varma N, Mathews V, Bhattacharyya J. et al. Myeloproliferative neoplasms working group consensus recommendations for diagnosis and management of primary myelofibrosis, polycythemia vera, and essential thrombocythemia. Indian J Med Paediatr Oncol 2015; 36: 3-16
- James C, Delhommeau F, Marzac C, Teyssandier I, Couédic JP, Giraudier S. et al. Detection of JAK2 V617F as a first intention diagnostic test for erythrocytosis. Leukemia 2006; 20: 350-3
- Pardanani A, Lasho TL, Finke C, Hanson CA, Tefferi A. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leuk 2007; 21: 1960-3
- Barbui T, Thiele J, Passamonti F, Rumi E, Boveri E, Ruggeri M. et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: An international study. J Clin Oncol 2011; 29: 3179-84
- Gong JZ, Cook JR, Greiner TC, Hedvat C, Hill CE, Lim MS. et al. Laboratory practice guidelines for detecting and reporting JAK2 and MPL mutations in myeloproliferative neoplasms: A report of the Association for Molecular Pathology. J Mol Diagn 2013; 15: 733-44
- Mesa RA. The evolving treatment paradigm in myelofibrosis. Leuk Lymphoma 2013; 54: 242-51
- Andersen CL, Andreasson B, Hasselbalch H, Hultcrantz M, Knutsen H, Lindgren M. et al. Nordic Guidelines On the Diagnosis and Treatment of Patients With Myeloproliferative Neoplasms. Available from: http://www.nmpnorg/indexphp/guidelines/1-nmpn-guidelines-2013/file2013. [Last accessed on 2017 Oct 17].
- Cervantes F. How I treat myelofibrosis. Blood 2014; 124: 2635-42
- ;Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E. et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009; 113: 2895-901
- Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S. et al. DIPSS plus: A refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol 2011; 29: 392-7
- Passamonti F, Cervantes F, Vannucchi AM, Morra E, Rumi E, Pereira A. et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010; 115: 1703-8
- Abelsson J, Merup M, Birgegård G, WeisBjerrum O, Brinch L, Brune M. et al. The outcome of allo-HSCT for 92 patients with myelofibrosis in the Nordic countries. Bone Marrow Transplant 2012; 47: 380-6
- Huang J, Tefferi A. Erythropoiesis stimulating agents have limited therapeutic activity in transfusion-dependent patients with primary myelofibrosis regardless of serum erythropoietin level. Eur J Haematol 2009; 83: 154-5
- Al-Ali HK, Stalbovskaya V, Gopalakrishna P. Ruxolitinib overcomes the adverse prognostic effect of anemia in patients with myelofibrosis. (abstract 4583). Blood 2014; 124: 4583
- Cervantes F, Hernández-Boluda JC, Alvarez A, Nadal E, Montserrat E. Danazol treatment of idiopathic myelofibrosis with severe anemia. Haematologica 2000; 85: 595-9
- Mesa RA, Tefferi A. Emerging drugs for the therapy of primary and post essential thrombocythemia, post polycythemia vera myelofibrosis. Expert Opin Emerg Drugs 2009; 14: 471-9
- Mesa RA, Steensma DP, Pardanani A, Li CY, Elliott M, Kaufmann SH. et al. A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia. Blood 2003; 101: 2534-41
- Löfvenberg E, Wahlin A. Management of polycythaemia vera, essential thrombocythaemia and myelofibrosis with hydroxyurea. Eur J Haematol 1988; 41: 375-81
- Manoharan A. Management of myelofibrosis with intermittent hydroxyurea. Br J Haematol 1991; 77: 252-4
- Martínez-Trillos A, Gaya A, Maffioli M, Arellano-Rodrigo E, Calvo X, Díaz-Beyá M. et al. Efficacy and tolerability of hydroxyurea in the treatment of the hyperproliferative manifestations of myelofibrosis: Results in 40 patients. Ann Hematol 2010; 89: 1233-7
- Jakavi. Package Insert. Mumbai, India: Novartis India Limited; 20 December 2016
- Harrison CN, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Gisslinger H, Knoops L. et al. Long-term efficacy and safety in COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for the treatment of myelofibrosis: 5-year final study results. Leukemia 2016; 30: 1701-7
- Kvasnicka HM, Thiele J, Bueso-Ramos CE, Sun W, Naim A, Sivaraman S. et al. Effects of Long-Term Ruxolitinib (RUX) on Bone Marrow (BM) Morphology in Patients with Myelofibrosis (MF) Enrolled in the COMFORT-I Study. Poster 1949. Presented at the American Society of Hematology Annual Meeting; 2016.
- Verstovsek S, Gupta V, Gotlib JR, Mesa RA, Vannucchi AM, Kiladjian JJ. et al. A Pooled Overall Survival Analysis of 5-Year Data from the COMFORT-I and COMFORT-II Trials of Ruxolitinib for the Treatment of Myelofibrosis. Poster 1949. Presented at the American Society of Hematology Annual Meeting; 2016
- Mesa R, Jamieson C, Bhatia R, Deininger MW, Gerds AT, Ivana G. et al. Myeloproliferative Neoplasms. National Comprehensive Cancer Network. Version I.2017; 2016.
- Latagliata R, Di Veroli A, Palumbo GA, Bonifacio M, Andriani A, Polverelli N. et al. Efficacy and Safety of Ruxolitinib in Elderly Patients (> 75 years) with Myelofibrosis. Abstract 4251. Presented at the American Society of Hematology Annual Meeting; 2016.
- Barosi G, Tefferi A, Besses C, Birgegard G, Cervantes F, Finazzi G. et al. Clinical end points for drug treatment trials in BCR-ABL1-negative classic myeloproliferative neoplasms: Consensus statements from European Leukemia Net (ELN) and Internation Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT). Leukemia 2015; 29: 20-6
- Tefferi A, Cervantes F, Mesa R, Passamonti F, Verstovsek S, Vannucchi AM. et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European Leukemia Net (ELN) consensus report. Blood 2013; 122: 1395-8
- Marchioli R, Finazzi G, Specchia G, Masciulli A, Mennitto MR, Barbui T. et al. The CYTO-PV: A large-scale trial testing the intensity of CYTOreductive therapy to prevent cardiovascular events in patients with polycythemia vera. Thrombosis 2011; 2011: 794240
- Barbui T, Thiele J, Vannucchi AM, Tefferi A. Rethinking the diagnostic criteria of polycythemia vera. Leukemia 2014; 28: 1191-5
- Griesshammer M, Gisslinger H, Mesa R. Current and future treatment options for polycythemia vera. Ann Hematol 2015; 94: 901-10
- Tibes R, Mesa RA. Emerging drugs for polycythemia vera. Expert Opin Emerg Drugs 2013; 18: 393-404
- Landolfi R, Marchioli R, Kutti J, Gisslinger H, Tognoni G, Patrono C. et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004; 350: 114-24
- Barosi G, Birgegard G, Finazzi G, Griesshammer M, Harrison C, Hasselbalch H. et al. A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: Results of a European Leukemia Net (ELN) consensus process. Br J Haematol 2010; 148: 961-3
- Barbui T, Finazzi MC, Finazzi G. Front-line therapy in polycythemia vera and essential thrombocythemia. Blood Rev 2012; 26: 205-11
- Verstovsek S, Vannucchi AM, Griesshammer M, Masszi T, Durrant S, Passamonti F. et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016; 101: 821-9
- Passamonti F, Griesshammer M, Palandri F, Egyed M, Benevolo G, Devos T. et al. Ruxolitinib proves superior to best available therapy in patients with polycythemia vera (PV) and a nonpalpable spleen: Results from the phase IIIb RESPONSE-2 study. Abstract Release date EHA Learning Center; May 19, 2016; Passamonti F. Label: Abstracts 2016; 10 Jun, 2016. p. 135145.
- ;Ballen KK, Shrestha S, Sobocinski KA, Zhang MJ, Bashey A, Bolwell BJ. et al. Outcome of transplantation for myelofibrosis. Biol Blood Marrow Transplant 2010; 16: 358-67
- Barbui T, Barosi G, Grossi A, Gugliotta L, Liberato LN, Marchetti M. et al. Practice guidelines for the therapy of essential thrombocythemia. A statement from the Italian Society of Hematology, the Italian Society of Experimental Hematology and the Italian Group for Bone Marrow Transplantation. Haematologica 2004; 89: 215-32
- Budde U, Schaefer G, Mueller N, Egli H, Dent J, Ruggeri Z. et al. Acquired von Willebrand’s disease in the myeloproliferative syndrome. Blood 1984; 64: 981-5
- Greist A. The role of blood component removal in essential and reactive thrombocytosis. Ther Apher 2002; 6: 36-44
- Björkholm M, Derolf AR, Hultcrantz M, Kristinsson SY, Ekstrand C, Goldin LR. et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol 2011; 29: 2410-5
- Kundranda MN, Tibes R, Mesa RA. Transformation of a chronic myeloproliferative neoplasm to acute myelogenous leukemia: Does anything work?. Curr Hematol Malig Rep 2012; 7: 78-86
- Thepot S, Itzykson R, Seegers V, Raffoux E, Quesnel B, Chait Y. et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: A report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 2010; 116: 3735-42
- Smalberg JH, Arends LR, Valla DC, Kiladjian JJ, Janssen HL, Leebeek FW. et al. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: A meta-analysis. Blood 2012; 120: 4921-8
- Griesshammer M, Struve S, Harrison CM. Essential thrombocythemia/polycythemia vera and pregnancy: The need for an observational study in Europe. Semin Thromb Hemost 2006; 32: 422-9
- Harrison CN, Robinson SE. Myeloproliferative disorders in pregnancy. Hematol Oncol Clin North Am 2011; 25: 261-75, vii
- Barbui T, Finazzi G. Special issues in myeloproliferative neoplasms. Curr Hematol Malig Rep 2011; 6: 28-35
Address for correspondence
Publication History
Article published online:
17 June 2021
© 2018. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2,
Noida-201301 UP, India
| Figure 1:Treatment algorithm for primary myelofibrosis
| Figure.2:Algorithm for management of essential thrombocythemia low-grade glioma subtypes
| Figure.3:Algorithm for management of polycythemia vera different low-grade glioma subtypes
References
- Levine RL, Gilliland DG. Myeloproliferative disorders. Blood 2008; 112: 2190-8
- Mascarenhas J, Mughal TI, Verstovsek S. Biology and clinical management of myeloproliferative neoplasms and development of the JAK inhibitor ruxolitinib. Curr Med Chem 2012; 19: 4399-413
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127: 2391-405
- Tefferi A. Essential thrombocythemia, polycythemia vera, and myelofibrosis: Current management and the prospect of targeted therapy. Am J Hematol 2008; 83: 491-7
- Cervantes F, Tassies D, Salgado C, Rovira M, Pereira A, Rozman C. et al. Acute transformation in nonleukemic chronic myeloproliferative disorders: Actuarial probability and main characteristics in a series of 218 patients. Acta Haematol 1991; 85: 124-7
- Visser O, Trama A, Maynadié M, Stiller C, Marcos-Gragera R, De Angelis R. et al. Incidence, survival and prevalence of myeloid malignancies in Europe. Eur J Cancer 2012; 48: 3257-66
- Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma 2014; 55: 595-600
- Moulard O, Mehta J, Fryzek J, Olivares R, Iqbal U, Mesa RA. et al. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol 2014; 92: 289-97
- Campbell PJ, Green AR. The myeloproliferative disorders. N Engl J Med 2006; 355: 2452-66
- ;Campbell PJ, Scott LM, Buck G, Wheatley K, East CL, Marsden JT. et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: A prospective study. Lancet 2005; 366: 1945-53
- Guglielmelli P, Lasho TL, Rotunno G, Score J, Mannarelli C, Pancrazzi A. et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: An international study of 797 patients. Leukemia 2014; 28: 1804-10
- Tefferi A, Guglielmelli P, Lasho TL, Rotunno G, Finke C, Mannarelli C. et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: An international study of 570 patients. Leukemia 2014; 28: 1494-500
- Agarwal MB, Malhotra H, Chakrabarti P, Varma N, Mathews V, Bhattacharyya J. et al. Myeloproliferative neoplasms working group consensus recommendations for diagnosis and management of primary myelofibrosis, polycythemia vera, and essential thrombocythemia. Indian J Med Paediatr Oncol 2015; 36: 3-16
- James C, Delhommeau F, Marzac C, Teyssandier I, Couédic JP, Giraudier S. et al. Detection of JAK2 V617F as a first intention diagnostic test for erythrocytosis. Leukemia 2006; 20: 350-3
- Pardanani A, Lasho TL, Finke C, Hanson CA, Tefferi A. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leuk 2007; 21: 1960-3
- Barbui T, Thiele J, Passamonti F, Rumi E, Boveri E, Ruggeri M. et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: An international study. J Clin Oncol 2011; 29: 3179-84
- Gong JZ, Cook JR, Greiner TC, Hedvat C, Hill CE, Lim MS. et al. Laboratory practice guidelines for detecting and reporting JAK2 and MPL mutations in myeloproliferative neoplasms: A report of the Association for Molecular Pathology. J Mol Diagn 2013; 15: 733-44
- Mesa RA. The evolving treatment paradigm in myelofibrosis. Leuk Lymphoma 2013; 54: 242-51
- Andersen CL, Andreasson B, Hasselbalch H, Hultcrantz M, Knutsen H, Lindgren M. et al. Nordic Guidelines On the Diagnosis and Treatment of Patients With Myeloproliferative Neoplasms. Available from: http://www.nmpnorg/indexphp/guidelines/1-nmpn-guidelines-2013/file2013. [Last accessed on 2017 Oct 17].
- Cervantes F. How I treat myelofibrosis. Blood 2014; 124: 2635-42
- ;Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E. et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009; 113: 2895-901
- Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S. et al. DIPSS plus: A refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol 2011; 29: 392-7
- Passamonti F, Cervantes F, Vannucchi AM, Morra E, Rumi E, Pereira A. et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010; 115: 1703-8
- Abelsson J, Merup M, Birgegård G, WeisBjerrum O, Brinch L, Brune M. et al. The outcome of allo-HSCT for 92 patients with myelofibrosis in the Nordic countries. Bone Marrow Transplant 2012; 47: 380-6
- Huang J, Tefferi A. Erythropoiesis stimulating agents have limited therapeutic activity in transfusion-dependent patients with primary myelofibrosis regardless of serum erythropoietin level. Eur J Haematol 2009; 83: 154-5
- Al-Ali HK, Stalbovskaya V, Gopalakrishna P. Ruxolitinib overcomes the adverse prognostic effect of anemia in patients with myelofibrosis. (abstract 4583). Blood 2014; 124: 4583
- Cervantes F, Hernández-Boluda JC, Alvarez A, Nadal E, Montserrat E. Danazol treatment of idiopathic myelofibrosis with severe anemia. Haematologica 2000; 85: 595-9
- Mesa RA, Tefferi A. Emerging drugs for the therapy of primary and post essential thrombocythemia, post polycythemia vera myelofibrosis. Expert Opin Emerg Drugs 2009; 14: 471-9
- Mesa RA, Steensma DP, Pardanani A, Li CY, Elliott M, Kaufmann SH. et al. A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia. Blood 2003; 101: 2534-41
- Löfvenberg E, Wahlin A. Management of polycythaemia vera, essential thrombocythaemia and myelofibrosis with hydroxyurea. Eur J Haematol 1988; 41: 375-81
- Manoharan A. Management of myelofibrosis with intermittent hydroxyurea. Br J Haematol 1991; 77: 252-4
- Martínez-Trillos A, Gaya A, Maffioli M, Arellano-Rodrigo E, Calvo X, Díaz-Beyá M. et al. Efficacy and tolerability of hydroxyurea in the treatment of the hyperproliferative manifestations of myelofibrosis: Results in 40 patients. Ann Hematol 2010; 89: 1233-7
- Jakavi. Package Insert. Mumbai, India: Novartis India Limited; 20 December 2016
- Harrison CN, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Gisslinger H, Knoops L. et al. Long-term efficacy and safety in COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for the treatment of myelofibrosis: 5-year final study results. Leukemia 2016; 30: 1701-7
- Kvasnicka HM, Thiele J, Bueso-Ramos CE, Sun W, Naim A, Sivaraman S. et al. Effects of Long-Term Ruxolitinib (RUX) on Bone Marrow (BM) Morphology in Patients with Myelofibrosis (MF) Enrolled in the COMFORT-I Study. Poster 1949. Presented at the American Society of Hematology Annual Meeting; 2016.
- Verstovsek S, Gupta V, Gotlib JR, Mesa RA, Vannucchi AM, Kiladjian JJ. et al. A Pooled Overall Survival Analysis of 5-Year Data from the COMFORT-I and COMFORT-II Trials of Ruxolitinib for the Treatment of Myelofibrosis. Poster 1949. Presented at the American Society of Hematology Annual Meeting; 2016
- Mesa R, Jamieson C, Bhatia R, Deininger MW, Gerds AT, Ivana G. et al. Myeloproliferative Neoplasms. National Comprehensive Cancer Network. Version I.2017; 2016.
- Latagliata R, Di Veroli A, Palumbo GA, Bonifacio M, Andriani A, Polverelli N. et al. Efficacy and Safety of Ruxolitinib in Elderly Patients (> 75 years) with Myelofibrosis. Abstract 4251. Presented at the American Society of Hematology Annual Meeting; 2016.
- Barosi G, Tefferi A, Besses C, Birgegard G, Cervantes F, Finazzi G. et al. Clinical end points for drug treatment trials in BCR-ABL1-negative classic myeloproliferative neoplasms: Consensus statements from European Leukemia Net (ELN) and Internation Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT). Leukemia 2015; 29: 20-6
- Tefferi A, Cervantes F, Mesa R, Passamonti F, Verstovsek S, Vannucchi AM. et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European Leukemia Net (ELN) consensus report. Blood 2013; 122: 1395-8
- Marchioli R, Finazzi G, Specchia G, Masciulli A, Mennitto MR, Barbui T. et al. The CYTO-PV: A large-scale trial testing the intensity of CYTOreductive therapy to prevent cardiovascular events in patients with polycythemia vera. Thrombosis 2011; 2011: 794240
- Barbui T, Thiele J, Vannucchi AM, Tefferi A. Rethinking the diagnostic criteria of polycythemia vera. Leukemia 2014; 28: 1191-5
- Griesshammer M, Gisslinger H, Mesa R. Current and future treatment options for polycythemia vera. Ann Hematol 2015; 94: 901-10
- Tibes R, Mesa RA. Emerging drugs for polycythemia vera. Expert Opin Emerg Drugs 2013; 18: 393-404
- Landolfi R, Marchioli R, Kutti J, Gisslinger H, Tognoni G, Patrono C. et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004; 350: 114-24
- Barosi G, Birgegard G, Finazzi G, Griesshammer M, Harrison C, Hasselbalch H. et al. A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: Results of a European Leukemia Net (ELN) consensus process. Br J Haematol 2010; 148: 961-3
- Barbui T, Finazzi MC, Finazzi G. Front-line therapy in polycythemia vera and essential thrombocythemia. Blood Rev 2012; 26: 205-11
- Verstovsek S, Vannucchi AM, Griesshammer M, Masszi T, Durrant S, Passamonti F. et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016; 101: 821-9
- Passamonti F, Griesshammer M, Palandri F, Egyed M, Benevolo G, Devos T. et al. Ruxolitinib proves superior to best available therapy in patients with polycythemia vera (PV) and a nonpalpable spleen: Results from the phase IIIb RESPONSE-2 study. Abstract Release date EHA Learning Center; May 19, 2016; Passamonti F. Label: Abstracts 2016; 10 Jun, 2016. p. 135145.
- ;Ballen KK, Shrestha S, Sobocinski KA, Zhang MJ, Bashey A, Bolwell BJ. et al. Outcome of transplantation for myelofibrosis. Biol Blood Marrow Transplant 2010; 16: 358-67
- Barbui T, Barosi G, Grossi A, Gugliotta L, Liberato LN, Marchetti M. et al. Practice guidelines for the therapy of essential thrombocythemia. A statement from the Italian Society of Hematology, the Italian Society of Experimental Hematology and the Italian Group for Bone Marrow Transplantation. Haematologica 2004; 89: 215-32
- Budde U, Schaefer G, Mueller N, Egli H, Dent J, Ruggeri Z. et al. Acquired von Willebrand’s disease in the myeloproliferative syndrome. Blood 1984; 64: 981-5
- Greist A. The role of blood component removal in essential and reactive thrombocytosis. Ther Apher 2002; 6: 36-44
- Björkholm M, Derolf AR, Hultcrantz M, Kristinsson SY, Ekstrand C, Goldin LR. et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol 2011; 29: 2410-5
- Kundranda MN, Tibes R, Mesa RA. Transformation of a chronic myeloproliferative neoplasm to acute myelogenous leukemia: Does anything work?. Curr Hematol Malig Rep 2012; 7: 78-86
- Thepot S, Itzykson R, Seegers V, Raffoux E, Quesnel B, Chait Y. et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: A report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 2010; 116: 3735-42
- Smalberg JH, Arends LR, Valla DC, Kiladjian JJ, Janssen HL, Leebeek FW. et al. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: A meta-analysis. Blood 2012; 120: 4921-8
- Griesshammer M, Struve S, Harrison CM. Essential thrombocythemia/polycythemia vera and pregnancy: The need for an observational study in Europe. Semin Thromb Hemost 2006; 32: 422-9
- Harrison CN, Robinson SE. Myeloproliferative disorders in pregnancy. Hematol Oncol Clin North Am 2011; 25: 261-75, vii
- Barbui T, Finazzi G. Special issues in myeloproliferative neoplasms. Curr Hematol Malig Rep 2011; 6: 28-35