 PDF
PDF  Views
Views  Share
Share


The “Blast” Behind Jerky Eyes
CC BY 4.0 · Indian J Med Paediatr Oncol 2023; 44(03): 353-355
DOI: DOI: 10.1055/s-0043-1761264
Abstract
Opsoclonus is defined as hyperkinetic, omnidirectional, spontaneous, and involuntary chaotic eye movements. Opsoclonus-myoclonus-ataxia syndrome is addressed by many names including “dancing eyes-dancing feet syndrome,” “Kinsbourne syndrome,” and “infantile polymyoclonia.” The early accounts of the clinical syndrome date back to 1962 when Marcel Kinsbourne described six cases of this phenotype. However, it was not until 1968 the association with occult neuroblastoma was first reported. We report the video of a 1-year-old boy who presented with this syndrome for a duration of 3 months. He was diagnosed to have an abdominal neuroblastoma and was treated with resection of the tumor and administration of intramuscular adrenocorticotropic hormone. He showed complete resolution of symptoms. The syndrome is difficult to recognize and might be confused with seizures, tremors, or chorea; hence, it is important that residents learnt to recognize this syndrome and look for an underlying tumor actively.
Publication History
Article published online:
12 May 2023
© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Opsoclonus is defined as hyperkinetic, omnidirectional, spontaneous, and involuntary chaotic eye movements. Opsoclonus-myoclonus-ataxia syndrome is addressed by many names including “dancing eyes-dancing feet syndrome,” “Kinsbourne syndrome,” and “infantile polymyoclonia.” The early accounts of the clinical syndrome date back to 1962 when Marcel Kinsbourne described six cases of this phenotype. However, it was not until 1968 the association with occult neuroblastoma was first reported. We report the video of a 1-year-old boy who presented with this syndrome for a duration of 3 months. He was diagnosed to have an abdominal neuroblastoma and was treated with resection of the tumor and administration of intramuscular adrenocorticotropic hormone. He showed complete resolution of symptoms. The syndrome is difficult to recognize and might be confused with seizures, tremors, or chorea; hence, it is important that residents learnt to recognize this syndrome and look for an underlying tumor actively.
Keywords
neuroblastoma - paraneoplastic syndrome - opsoclonus-myoclonus-ataxia syndromeCase Description
A 1-year-old boy presented with complaints of regression of milestones and tremulousness of neck for a duration of 3 months. He was born out of nonconsanguineous marriage at term with an uneventful perinatal period. The neurodevelopment was normal (sitting without support, speaking bisyllables and having stranger anxiety) till 8 months of age. After this age, he gradually lost milestones and now he was unable to speak any words or sit even with support. As per parents, the child was excessively irritable and slept for very short durations. On examination, vitals were stable, no significant pallor, icterus, neurocutaneous markers, apparent congenital malformation, abnormal odor, or organomegaly were noted. There were opsoclonus movements of eyes in all directions of gaze; these movements also persisted during sleep. The truncal instability was there along with intermittent jerky movements of neck and limbs. No cranial nerve palsy, tone abnormality or focal neurological deficit was noted ([Video 1]). Rest of the systemic examination was normal.
Video 1 Video of the child showing opsoclonus movement of eyes in all direction with tremulousness of head.
<video width="320" height="240" controls="">In view of the clinical picture, the possibility of opsoclonus-myoclonus-ataxia syndrome (OMAS) was considered. The electroencephalography revealed no evidence of seizure activity. Magnetic resonance imaging of brain was grossly normal. Chest X-ray and ultrasound abdomen showed no mass. Cerebrospinal fluid analysis revealed acellular fluid with sugar and protein of 70 and 20 mg/dL, respectively. Contrast-enhanced computed tomography (CECT) of abdomen revealed a large heterogeneously enhancing lesion measuring 2.5 × 3.1 × 4.5 cm in the retroperitoneum involving organ of Zuckerkandl ([Fig. 1A and B]). This was followed by a fluorine 18 fluorodeoxyglucose single-photon emission computerized tomography that showed increased tracer uptake (maximum standardized uptake value: 6.9, 4.6 × 2.6 × 4.8 cm) in the retroperitoneal soft tissue mass implicating somatostatin receptor expressing retroperitoneal soft tissue mass. Due to financial constraints, urine catecholamine metabolites were not assessed.
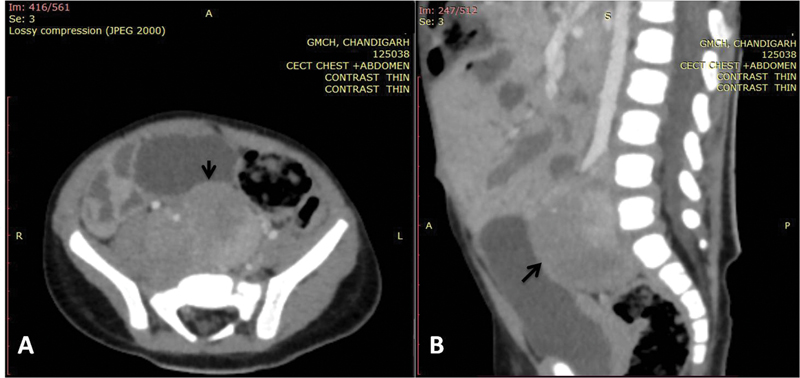
| Fig 1 :(A) Axial and (B) sagittal image of contrast-enhanced computed tomogram of abdomen showing well-defined heterogeneously enhancing mass lesion (black arrows) in the retroperitoneum, in the midline and on the right side, extending cranially from the level of aortic bifurcation and caudally into the rectovesical pouch in the pelvis till S2-S3 vertebral level displacing the right iliac vessels anterolaterally.
The patient was referred to higher center for further management. The mass was surgically excised. The histopathology of excised mass was suggestive of ganglioneuroblastoma, intermixed subtype. Child was started on pulsed dexamethasone (20 mg/m2/day in two divided doses for 3 consecutive days every month), intramuscular adrenocorticotropic hormone (ACTH), and monthly intravenous immunoglobulin (IVIG) (1–2 g/kg every 4 weeks for 12 months). IV cyclophosphamide (25 mg/kg) was given at 4 weekly intervals for three doses. In view of partial response at 3 months, therapy was escalated to rituximab (375 mg/m2/dose once a week for 4 doses). Child is currently asymptomatic and has regained regressed milestones. At 18 months follow-up, child has neither experienced recurrence of symptoms nor shown any evidence of tumor on repeat imaging.
Discussion
The early accounts of clinical syndrome of OMAS date back to 1962 when Marcel Kinsbourne described six cases of this phenotype. However, it was not until 1968 the association with occult neuroblastoma was first reported. Studies in India have demonstrated that this entity contributes to 7% children presenting with movement disorders.[1] [2] The syndrome might be confused with seizures, tremors, or chorea. In most of the cases, the etiology is paraneoplastic. However, parainfectious etiology has also been seen. Around 50% of pediatric cases of OMAS are accounted for by an underlying neuroblastoma, whereas only 2 to 3% of neuroblastomas present with this paraneoplastic syndrome.[3] [4] The pathophysiology of this disorder remains an enigma. There have been speculations about damage to omnipause cells in the nucleus raphe interpositus of pons and disinhibition of fastigial nucleus in cerebellum.[5] The diagnosis is primarily clinical, but the search for the underlying cause needs to be extensive and thorough with CECT chest, abdomen, and pelvis actively looking for a tumor. Neuroblastoma is the most common underlying tumor (73%) that is followed by ganglioneuroblastoma (22%) and ganglioneuroma (4%) in that order.[6] The management strategy is two pronged: management of underlying tumor and immunotherapy for OMAS. The tumor is treated with resection and/or chemotherapy depending upon risk stratification. As for the paraneoplastic manifestation, either an aggressive upfront approach or gradual escalation approach may be followed. All regimens involve the use of steroids (prednisolone, ACTH, or dexamethasone pulses), IVIG, cyclophosphamide, and rituximab. Resistant cases might require plasmapheresis.[7] OMAS-associated neuroblastic tumors have shown favorable outcomes when compared with those without OMAS.[3] It has been seen that with an increase in the pre-treatment duration, the prognosis worsens and the risk of long-term neurological sequelae increases.[4] Thus, early diagnosis and management are important to improve prognosis. Also, it is noteworthy that patients in OMAS in whom no tumor is localized after adequate investigation, the search for tumor must be repeated at 6 months as small sized tumors might be missed in the initial screening.
Conflict of Interest
None declared.
Informed Consent
A written informed consent was taken from the parents of the child.
References
- Goraya JS. Acute movement disorders in children: experience from a developing country. J Child Neurol 2015; 30 (04) 406-411
- Neurological sequelae of the dancing eye syndrome - PubMed [Internet]. [cited 2021 Mar 19]. Accessed January 8, 2023, at: https://pubmed.ncbi.nlm.nih.gov/8929735/
- Sahu JK, Prasad K. The opsoclonus–myoclonus syndrome. Pract Neurol 2011; 11 (03) 160-166
- Galstyan A, Wilbur C, Selby K, Hukin J. Opsoclonus-myoclonus syndrome: a new era of improved prognosis?. Pediatr Neurol 2017; 72: 65-69
- Pranzatelli MR. The neurobiology of the opsoclonus-myoclonus syndrome. Clin Neuropharmacol 1992; 15 (03) 186-228
- Pranzatelli MR, Tate ED, McGee NR. Demographic, clinical, and immunologic features of 389 children with opsoclonus-myoclonus syndrome: a cross-sectional study. Front Neurol 2017; 8: 468 https://www.frontiersin.org/articles/10.3389/fneur.2017.00468 cited 2022Oct26 [Internet]
- Rossor T, Yeh EA, Khakoo Y. et al; OMS Study Group. Diagnosis and management of opsoclonus-myoclonus-ataxia syndrome in children: an international perspective. Neurol Neuroimmunol Neuroinflamm 2022; 9 (03) e1153 DOI: 10.1212/NXI.0000000000001153.
Address for correspondence
Chandrika Azad, MDDepartment of Pediatrics, Government Medical College and HospitalSector 32, Chandigarh, 160036IndiaEmail: chandrika_azad@yahoo.co.inPublication History
Article published online:
12 May 2023© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Fig 1 :(A) Axial and (B) sagittal image of contrast-enhanced computed tomogram of abdomen showing well-defined heterogeneously enhancing mass lesion (black arrows) in the retroperitoneum, in the midline and on the right side, extending cranially from the level of aortic bifurcation and caudally into the rectovesical pouch in the pelvis till S2-S3 vertebral level displacing the right iliac vessels anterolaterally.
References
- Goraya JS. Acute movement disorders in children: experience from a developing country. J Child Neurol 2015; 30 (04) 406-411
- Neurological sequelae of the dancing eye syndrome - PubMed [Internet]. [cited 2021 Mar 19]. Accessed January 8, 2023, at: https://pubmed.ncbi.nlm.nih.gov/8929735/
- Sahu JK, Prasad K. The opsoclonus–myoclonus syndrome. Pract Neurol 2011; 11 (03) 160-166
- Galstyan A, Wilbur C, Selby K, Hukin J. Opsoclonus-myoclonus syndrome: a new era of improved prognosis?. Pediatr Neurol 2017; 72: 65-69
- Pranzatelli MR. The neurobiology of the opsoclonus-myoclonus syndrome. Clin Neuropharmacol 1992; 15 (03) 186-228
- Pranzatelli MR, Tate ED, McGee NR. Demographic, clinical, and immunologic features of 389 children with opsoclonus-myoclonus syndrome: a cross-sectional study. Front Neurol 2017; 8: 468 https://www.frontiersin.org/articles/10.3389/fneur.2017.00468 cited 2022Oct26 [Internet]
- Rossor T, Yeh EA, Khakoo Y. et al; OMS Study Group. Diagnosis and management of opsoclonus-myoclonus-ataxia syndrome in children: an international perspective. Neurol Neuroimmunol Neuroinflamm 2022; 9 (03) e1153 DOI: 10.1212/NXI.0000000000001153.