 PDF
PDF  Views
Views  Share
Share


The Tumor Microenvironment
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2020; 41(02): 213-214
DOI: DOI: 10.4103/ijmpo.ijmpo_25_20
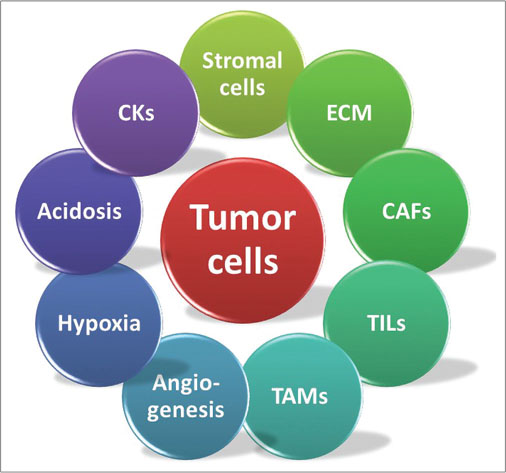
| Figure 1: The tumor and its microenvironment. ECM – Extracellular matrix; CAF – Cancer-associated fibroblast; TIL – Tumor-infiltrating lymphocyte; TAM – Tumor-associated macrophage; CK – Cytokines
The nonmalignant cells in the TME may originate from native tissue or may be recruited into the TME from the peripheral blood or the bone marrow. These include stromal cells, fibroblasts, immune cells (T-cells, B-cells, natural killer [NK]-cells, and tumor-associated macrophages [TAMs]), as well as pericytes and adipocytes. Cancer-associated fibroblasts (CAFs) have a significant role in sculpting the TME, especially in tumor migration and invasion. These are thought to originate from resident fibroblasts, mesenchymal stem cells (MSC), or endothelial cells and are induced by the tumor cells to produce cytokines (e.g., interleukin [IL]-6 and IL-8), chemokines (e.g., C-X-C motif ligand 1 [CXCL1]), growth factors (e.g., insulin-like growth factor 2), as well as components of the ECM. The ECM is a dynamic network of structural macromolecules, such as collagen, elastin, tenascin, laminin, and proteoglycans, which act as scaffolding for the cellular components of the TME. An abnormal ECM can promote inflammation, tumor angiogenesis, and abnormal functioning of the stromal cells. The importance of tumor angiogenesis was first hypothesized by the “Father of Angiogenesis” Judah Folkman as far back as 1971. Without it, tumors cannot grow beyond a volume of 2 mm3.[1] Their rapid growth quickly outgrows their blood supply, leading to a deprivation of oxygen and nutrients, which creates regions of hypoxia and acidosis within the tumor. However, the newly formed blood vessels tend to suffer from chaotic branching, irregular lumens, and increased permeability, giving rise to unequal blood flow to different areas within the same tumor and an uneven distribution of both nutrients and drugs. The tumor endothelial cells (TECs) that line them are also remarkable, bigger in size, with abnormal cytogenetics and an increased sensitivity to growth factors, that gives them an antiapoptotic phenotype. They kill T-cells, inhibit their recruitment and activation, and have been implicated in tumor resistance to both conventional chemotherapy and immunotherapy.
Paget propounded his “seed and soil” hypothesis in 1889, wherein he proposed that the progression and metastasis of cancer is not random but the result of elaborate interactions between the tumor (seed) and the TME (soil). We now know that TME plays a central role in tumor initiation, promotion, and progression, as can be seen from the following examples. Fibroblasts and stromal cells in the TME produce growth factors, such as transforming growth factor-β and fibroblast growth factor, which enable the multiplication, metastases, and maintenance of malignant cells, as well as the migration of immune cells into the TME. Heat shock factor 1 is a transcriptional regulator, which when activated in CAFs, encourages epithelial–mesenchymal transition and progression to malignancy, while CXCL12 from MSCs, osteoblasts, or CAR (CXCL12-abundant reticular) cells mediates cross-talk between leukemic cells and the TME, enabling leukemic stem cells to settle into their bone marrow niche. Immune cells can both inhibit and promote tumorigenesis. Established tumors have an immunosuppressive microenvironment which blocks antitumor immunity. CD8+ memory T-cells, CD4+ T-helper 1, and interferon (IFN)-γ help in suppressing the development of cancers, whereas infiltrating B-cells in the TME are known to be protumorigenic. The presence of NK and NK T-cells in the TME is associated with a good prognosis in many tumors. Among the myeloid lineage cells found in the TME, myeloid-derived suppressor cells and mast cells help to promote tumor development. TAMs found in the TME derive from monocytes. TAMs can broadly be divided into two types, M1 and M2. M1 macrophages are “classically” activated by IFN-γ, along with either lipopolysaccharide or tumor necrosis factor. They are pro-inflammatory and antitumorigenic. M2 macrophages, on the other hand, undergo “alternative” activation by IL-4 and are anti-inflammatory and protumorigenic. TAMs are more abundant in hypoxic regions within the tumor mass and promote angiogenesis, immune dysregulation, tumor cell migration, invasion, metastasis, and chemoresistance.
Not only does the TME contribute significantly to chemoresistance, the TMEs of different tumors have also been found to share certain similarities such that the composition of a tumor’s TME can be used as a prognostic marker. For instance, pancreatic ductal adenocarcinomas (PDACs) are known for the complexity of their TMEs. Most PDACs (>50%) display an “immune-escape” phenotype, meaning an immunosuppressive TME replete with FOXP3+ Tregs and M2 TAMs, resulting in an aggressive clinical course and a median overall survival (OS) of 10 months.[2] On the other hand, those with an “immune-rich” TME have an abundance of T-cells, B-cells, and M1 TAMs; more favorable clinicopathological features; and better outcomes (median OS of 19–23 months). There is also an “immune-exhausted” poor-prognosis subtype comprising populations with high PD-L1 expression or microsatellite instability.
Research into the TME has also opened up new therapeutic avenues, enabling us to indirectly attacked tumors by modifying the TME, such as by altering the hypoxic milieu, the abnormal growth pathways, or the interactions between the tumor cells and the TME components. Among the approaches being studied in this regard are fibroblast activation protein inhibitors targeting the CAFs, CTLA-4 monoclonal antibodies targeting the immune system, kinase inhibitors targeting the signaling pathways, matrix metalloproteinase inhibitors targeting the ECM, and vascular endothelial growth factor inhibitors targeting tumor angiogenesis. Among the more established drugs, trabectedin is selectively cytotoxic for TAMs[3] and causes a significant downregulation of cytokines, chemokines, and inflammatory and angiogenic mediators, all of which add to its antitumor effects. Co-administration of the angiotensin inhibitor losartan has been found to sensitize pancreatic cancer to neoadjuvant FOLFIRINOX chemotherapy and improve outcomes by modifying the microenvironment, increasing drug and oxygen delivery to cancer cells, and activating immune pathways.[4] The nonselective β-blocker propranolol has been shown to have a high response rate in angiosarcomas and other vascular tumors. It decreases cancer cell viability by enhancing T-cell infiltration, decreasing PD1 expression, and inhibiting the proliferation and differentiation of TECs. The many successes of oral metronomic chemotherapies can be attributed to the normalization of tumor vasculature, inhibition of endothelial cell proliferation and migration, increased expression of the antiangiogenic protein thrombospondin-1, suppression of Tregs, functional restoration of NK cells and cytotoxic T-cells, and induction of dendritic cell maturation.
Publication History
Received: 27 January 2020
Accepted: 09 April 2020
Article published online:
23 May 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
The tumor microenvironment (TME) is the sum of all the hings the tumor cells are immersed in, including other nonmalignant cells, signaling molecules, the extracellular matrix (ECM), and the surrounding blood vessels. These components are involved in a number of intricate interactions with each other and with the cancer cells, as depicted in [Figure 1]. In 1986, HF Dworak described tumors as “wounds that do not heal,” alluding to the similarities between the TME and areas of chronic inflammation in tissues, both having rapidly growing cells causing local tissue ischemia which stimulates the growth of new blood vessels, and through them, an influx of immune cells. However, since a healing wound lacks both cancer stem cells and a limitless supply of growth factors, the healing process merely results in scar formation and not tumors.
| Figure 1: The tumor and its microenvironment. ECM – Extracellular matrix; CAF – Cancer-associated fibroblast; TIL – Tumor-infiltrating lymphocyte; TAM – Tumor-associated macrophage; CK – Cytokines
The nonmalignant cells in the TME may originate from native tissue or may be recruited into the TME from the peripheral blood or the bone marrow. These include stromal cells, fibroblasts, immune cells (T-cells, B-cells, natural killer [NK]-cells, and tumor-associated macrophages [TAMs]), as well as pericytes and adipocytes. Cancer-associated fibroblasts (CAFs) have a significant role in sculpting the TME, especially in tumor migration and invasion. These are thought to originate from resident fibroblasts, mesenchymal stem cells (MSC), or endothelial cells and are induced by the tumor cells to produce cytokines (e.g., interleukin [IL]-6 and IL-8), chemokines (e.g., C-X-C motif ligand 1 [CXCL1]), growth factors (e.g., insulin-like growth factor 2), as well as components of the ECM. The ECM is a dynamic network of structural macromolecules, such as collagen, elastin, tenascin, laminin, and proteoglycans, which act as scaffolding for the cellular components of the TME. An abnormal ECM can promote inflammation, tumor angiogenesis, and abnormal functioning of the stromal cells. The importance of tumor angiogenesis was first hypothesized by the “Father of Angiogenesis” Judah Folkman as far back as 1971. Without it, tumors cannot grow beyond a volume of 2 mm3.[1] Their rapid growth quickly outgrows their blood supply, leading to a deprivation of oxygen and nutrients, which creates regions of hypoxia and acidosis within the tumor. However, the newly formed blood vessels tend to suffer from chaotic branching, irregular lumens, and increased permeability, giving rise to unequal blood flow to different areas within the same tumor and an uneven distribution of both nutrients and drugs. The tumor endothelial cells (TECs) that line them are also remarkable, bigger in size, with abnormal cytogenetics and an increased sensitivity to growth factors, that gives them an antiapoptotic phenotype. They kill T-cells, inhibit their recruitment and activation, and have been implicated in tumor resistance to both conventional chemotherapy and immunotherapy.
Paget propounded his “seed and soil” hypothesis in 1889, wherein he proposed that the progression and metastasis of cancer is not random but the result of elaborate interactions between the tumor (seed) and the TME (soil). We now know that TME plays a central role in tumor initiation, promotion, and progression, as can be seen from the following examples. Fibroblasts and stromal cells in the TME produce growth factors, such as transforming growth factor-β and fibroblast growth factor, which enable the multiplication, metastases, and maintenance of malignant cells, as well as the migration of immune cells into the TME. Heat shock factor 1 is a transcriptional regulator, which when activated in CAFs, encourages epithelial–mesenchymal transition and progression to malignancy, while CXCL12 from MSCs, osteoblasts, or CAR (CXCL12-abundant reticular) cells mediates cross-talk between leukemic cells and the TME, enabling leukemic stem cells to settle into their bone marrow niche. Immune cells can both inhibit and promote tumorigenesis. Established tumors have an immunosuppressive microenvironment which blocks antitumor immunity. CD8+ memory T-cells, CD4+ T-helper 1, and interferon (IFN)-γ help in suppressing the development of cancers, whereas infiltrating B-cells in the TME are known to be protumorigenic. The presence of NK and NK T-cells in the TME is associated with a good prognosis in many tumors. Among the myeloid lineage cells found in the TME, myeloid-derived suppressor cells and mast cells help to promote tumor development. TAMs found in the TME derive from monocytes. TAMs can broadly be divided into two types, M1 and M2. M1 macrophages are “classically” activated by IFN-γ, along with either lipopolysaccharide or tumor necrosis factor. They are pro-inflammatory and antitumorigenic. M2 macrophages, on the other hand, undergo “alternative” activation by IL-4 and are anti-inflammatory and protumorigenic. TAMs are more abundant in hypoxic regions within the tumor mass and promote angiogenesis, immune dysregulation, tumor cell migration, invasion, metastasis, and chemoresistance.
Not only does the TME contribute significantly to chemoresistance, the TMEs of different tumors have also been found to share certain similarities such that the composition of a tumor’s TME can be used as a prognostic marker. For instance, pancreatic ductal adenocarcinomas (PDACs) are known for the complexity of their TMEs. Most PDACs (>50%) display an “immune-escape” phenotype, meaning an immunosuppressive TME replete with FOXP3+ Tregs and M2 TAMs, resulting in an aggressive clinical course and a median overall survival (OS) of 10 months.[2] On the other hand, those with an “immune-rich” TME have an abundance of T-cells, B-cells, and M1 TAMs; more favorable clinicopathological features; and better outcomes (median OS of 19–23 months). There is also an “immune-exhausted” poor-prognosis subtype comprising populations with high PD-L1 expression or microsatellite instability.
Research into the TME has also opened up new therapeutic avenues, enabling us to indirectly attacked tumors by modifying the TME, such as by altering the hypoxic milieu, the abnormal growth pathways, or the interactions between the tumor cells and the TME components. Among the approaches being studied in this regard are fibroblast activation protein inhibitors targeting the CAFs, CTLA-4 monoclonal antibodies targeting the immune system, kinase inhibitors targeting the signaling pathways, matrix metalloproteinase inhibitors targeting the ECM, and vascular endothelial growth factor inhibitors targeting tumor angiogenesis. Among the more established drugs, trabectedin is selectively cytotoxic for TAMs[3] and causes a significant downregulation of cytokines, chemokines, and inflammatory and angiogenic mediators, all of which add to its antitumor effects. Co-administration of the angiotensin inhibitor losartan has been found to sensitize pancreatic cancer to neoadjuvant FOLFIRINOX chemotherapy and improve outcomes by modifying the microenvironment, increasing drug and oxygen delivery to cancer cells, and activating immune pathways.[4] The nonselective β-blocker propranolol has been shown to have a high response rate in angiosarcomas and other vascular tumors. It decreases cancer cell viability by enhancing T-cell infiltration, decreasing PD1 expression, and inhibiting the proliferation and differentiation of TECs. The many successes of oral metronomic chemotherapies can be attributed to the normalization of tumor vasculature, inhibition of endothelial cell proliferation and migration, increased expression of the antiangiogenic protein thrombospondin-1, suppression of Tregs, functional restoration of NK cells and cytotoxic T-cells, and induction of dendritic cell maturation.
Conflict of Interest
There are no conflicts of interest.
References
- Folkman J, Cole P, Zimmerman S. Tumor behavior in isolated perfused organs: In vitro growth and metastases of biopsy material in rabbit thyroid and canine intestinal segment. Ann Surg 1966; 164: 491-502
- Wartenberg M, Cibin S, Zlobec I, Vassella E, Eppenberger-Castori S, Terracciano L. et al. Integrated genomic and immunophenotypic classification of pancreatic cancer reveals three distinct subtypes with prognostic/predictive significance. Clin Cancer Res 2018; 24: 4444-54
- Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M. et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013; 23: 249-62
- Murphy JE, Wo JY, Ryan DP, Clark JW, Jiang W, Yeap BY. et al. Total neoadjuvant therapy with FOLFIRINOX in combination with losartan followed by chemoradiotherapy for locally advanced pancreatic cancer: A phase 2 clinical trial. JAMA Oncol 2019; 5: 1020-7
Address for correspondence
Publication History
Received: 27 January 2020
Accepted: 09 April 2020
Article published online:
23 May 2021
© 2020. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
| Figure 1: The tumor and its microenvironment. ECM – Extracellular matrix; CAF – Cancer-associated fibroblast; TIL – Tumor-infiltrating lymphocyte; TAM – Tumor-associated macrophage; CK – Cytokines
- 1 Folkman J, Cole P, Zimmerman S. Tumor behavior in isolated perfused organs: In vitro growth and metastases of biopsy material in rabbit thyroid and canine intestinal segment. Ann Surg 1966; 164: 491-502
- 2 Wartenberg M, Cibin S, Zlobec I, Vassella E, Eppenberger-Castori S, Terracciano L. et al. Integrated genomic and immunophenotypic classification of pancreatic cancer reveals three distinct subtypes with prognostic/predictive significance. Clin Cancer Res 2018; 24: 4444-54
- 3 Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M. et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013; 23: 249-62
- 4 Murphy JE, Wo JY, Ryan DP, Clark JW, Jiang W, Yeap BY. et al. Total neoadjuvant therapy with FOLFIRINOX in combination with losartan followed by chemoradiotherapy for locally advanced pancreatic cancer: A phase 2 clinical trial. JAMA Oncol 2019; 5: 1020-7