 PDF
PDF  Views
Views  Share
Share


Thiopurine methyltransferase polymorphisms in children with acute lymphoblastic leukemia
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2014; 35(04): 276-280
DOI: DOI: 10.4103/0971-5851.144989
Abstract
Introduction: Acute lymphoblastic leukemia (ALL) is the most common malignancy in children. 6-mercaptopurine (6-MP) and methotrexate are backbone drugs for maintenance phase of treatment. Purine Analogs 6-MP/6-thioguanine/azathiopurine are metabolized to its inactive form by the enzyme thiopurine methyltransferase (TPMT). Ninety percent of the population harbor wild type on both alleles (TPMT wild/wild), 10% are heterozygous, that is, one allele is mutant (TPMT wild/mutant) and 0.3% are homozygous, that is, both allele are mutant (TPMT mutant/mutant). In heterozygous and homozygous variant, activity of enzyme is low, leading to a higher incidence of toxicity (myelosuppression). Aim: The primary objective was to access the polymorphism of the enzyme, TPMT, in Children with ALL. Secondary objective was to correlate TPMT genotype with 6-MP toxicities. Materials and Methods: Seventy-two children with newly diagnosed ALL during first maintenance phase were serially enrolled after obtaining consent. Five ml of peripheral blood was drawn and DNA extracted. TPMT 2 polymorphisms were performed using Allele specific polymerase chain reaction (PCR) and TPMT 3B and 3C are performed by PCR-restriction fragment length polymorphism. Results: Sixty-nine children of 72 (95.8%) were wild for TPMT polymorphism and 3 (4.2%) were heterozygous for TPMT. Among the heterozygous variant one each (33.3%) were heterozygous for 2A, 3A, 3C. Febrile neutropenia was the most common toxicity in both wild and heterozygous group. Conclusion: The frequency of TPMT polymorphisms in children with ALL is 4.2%. Heterozygous variant is this study are one each (33%) of 2A, 3A, 3C.
Publication History
Article published online:
19 July 2021
© 2014. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Introduction:
Acute lymphoblastic leukemia (ALL) is the most common malignancy in children. 6-mercaptopurine (6-MP) and methotrexate are backbone drugs for maintenance phase of treatment. Purine Analogs 6-MP/6-thioguanine/azathiopurine are metabolized to its inactive form by the enzyme thiopurine methyltransferase (TPMT). Ninety percent of the population harbor wild type on both alleles (TPMT wild/wild), 10% are heterozygous, that is, one allele is mutant (TPMT wild/mutant) and 0.3% are homozygous, that is, both allele are mutant (TPMT mutant/mutant). In heterozygous and homozygous variant, activity of enzyme is low, leading to a higher incidence of toxicity (myelosuppression).
Aim:
The primary objective was to access the polymorphism of the enzyme, TPMT, in Children with ALL. Secondary objective was to correlate TPMT genotype with 6-MP toxicities.
Materials and Methods:
Seventy-two children with newly diagnosed ALL during first maintenance phase were serially enrolled after obtaining consent. Five ml of peripheral blood was drawn and DNA extracted. TPMT 2 polymorphisms were performed using Allele specific polymerase chain reaction (PCR) and TPMT 3B and 3C are performed by PCR-restriction fragment length polymorphism.
Results:
Sixty-nine children of 72 (95.8%) were wild for TPMT polymorphism and 3 (4.2%) were heterozygous for TPMT. Among the heterozygous variant one each (33.3%) were heterozygous for 2A, 3A, 3C. Febrile neutropenia was the most common toxicity in both wild and heterozygous group.
Conclusion:
The frequency of TPMT polymorphisms in children with ALL is 4.2%. Heterozygous variant is this study are one each (33%) of 2A, 3A, 3C.
INTRODUCTION
Acute lymphoblastic leukemia (ALL) is the most common cancer in childhood.[1] 6-mercaptopurine (6-MP) and methotrexate (MTX) constitute the backbone for maintenance therapy. 6-MP is metabolized to its inactive form by an enzyme thiopurine methyltransferase (TPMT).[2] The enzyme levels are variable in the population. One in 300 persons has very low levels leading to severe toxicity due to 6-MP metabolites. 10% of population have intermediate level with varied toxicity and 90% of population has normal levels. There are 29 polymorphisms described for this enzyme of which TPMT 2, TPMT 3A, TPMT 3B, TPMT 3C (mutant) constitute 95% of polymorphisms.[3,4]
The single nucleotide polymorphisms are associated with lower TPMT activity, among which the most common are: 238 G > C transversion, 460 G > A transition, and 719 A > G transition. Three alleles account for more than 95% of the clinically relevant TPMT variants: TPMT * 2, TPMT * 3A, and TPMT * 3C. Wild-type has been designated as TPMT * 1. TPMT * 2 allele contains single 238 G > C mutation and TPMT * 3A allele has 2 mutations (460 G > A and 719 A > G), whereas TPMT * 3C has only 719 A > G mutation. A rare TPMT * 3B allele contains 460 G > A mutation only.
There are few studies from India reporting on TPMT gene polymorphisms in ALL patients[5,6,7,8] and others in healthy subjects.[9,10] Further studies in this field would help to elucidate the frequency of polymorphisms in Indian ALL population and toxicities with 6-MP.
The primary objective was to access the polymorphism of TPMT, in Children with ALL and the secondary objective was to correlate TPMT genotype with 6-MP toxicities.
MATERIALS AND METHODS
This is a prospective study conducted under the department of medical oncology, Nizam's Institute of Medical Sciences from September 2007 to January 2010. Institutional ethical approval was obtained. All the parents were informed about the study and consent was obtained. All newly diagnosed ALL children aged <18>
Methodology
5 ml of peripheral blood was drawn in ethylenediaminetetraacetic acid vaccutainers. Genomic DNA was extracted from peripheral leucocytes using the Qiagen DNA extraction kit. An allele-specific polymerase chain reaction (PCR) was used to analyze the TPMT * 2A (G238C) mutation. PCR amplification restriction fragment length polymorphism enzyme digestion were used to detect TPMT * 3B (G460A) and TPMT * 3C (A719G) mutations, as described previously.[11,12]
Study design
All the children were uniformly treated with the MCP-841 chemotherapy protocol as previously described.[13] Prerequisites before starting on chemotherapy were that the child had to be clinically stable with a total leukocyte count of more than 4000/cumm and platelet count of more than 100000/cumm. In MCP-841 protocol each maintenance phase contains a pulse chemotherapy phase (injectable) and 3 months of oral phase. The drugs delivered during injectable phase are vincristine, prednisolone, L-asparaginase, and daunomycin. In oral phase, oral 6-MP was started at dose of 75 mg/m2 /day for 3 weeks and oral MTX at a dose of 15 mg/m2 /weekly for 3 weeks followed by 1-week of drug holiday. Second and third oral phases were repeated as above.
As per the protocol, the dose intensity of 6-MP was 56.25 mg/m2 /day, and MTX 11.25 mg/m2 /week. As per the protocol, three oral phases took 12 weeks to complete.
During the 1st maintenance phase on oral 6-MP and MTX, the following toxicities were recorded as per National Cancer Institute-Common Toxicity Criteria Version 3: Febrile Neutropenia (FN) — patients with low risk (neutrophil count expected to recover within 5 days) with no focus of infection or hypotension were treated with oral antibiotics. Rest of all the children were treated as high risk with parenteral antibiotics; hepatotoxicity-Increase in serum transaminases and bilirubin; mucositis; number of weeks taken to complete first oral maintenance phase; the actual delivered dose of 6-MP and MTX against the intended dose intensity for 6-MP-56.25 mg/m2 /day and MTX-11.25 mg/m2 /week.
Dose interruption was defined as a child who was not on any medications for more than 2 weeks from the last dose of 6-MP. Dose adjustment was defined as any child requiring a dose reduction of 6-MP. Intolerance to drug was defined as patients having gastritis, nausea, and vomiting after taking 6-MP.
Statistical analysis: Statistical analysis was performed with SPSS 10.0 software (IBM corporation). A 2 × 2 test was used to compare frequency of alleles in studied population. 2 × 2 analysis was also used for testing the deviation of genotype proportions from Hardy-Weinberg equilibrium (HWE).
Patient characteristics were analyzed using descriptive statistic. Inferential statistics between two groups to calculate the P values were done using two tailored Fisher exact test using GraphPad Prism online calculator (GraphPad Software, Inc).
RESULTS
Seventy-two children with ALL treated consecutively were enrolled and toxicity was assessed. Median age for this group was 9 years (range, 2-18). The boy to girl ratio was 3.7:1. The median total leukocyte count at diagnosis was 10,800/mm3 (range, 1100-350000/mm3). Sixty-nine children (95.8%) were wild for TPMT polymorphism and 3 (4.2%) were heterozygous for TPMT. Among the heterozygous variant one each (33.3%) were heterozygous for 2A, 3A, 3C.
Allelic frequency of TPMT polymorphisms was out of 144 allele-141 (97.9%) allele were of wild type and one each 1/144 (0.69%) were 2A, 3A, 3C genotypes. These proportions followed HWE.
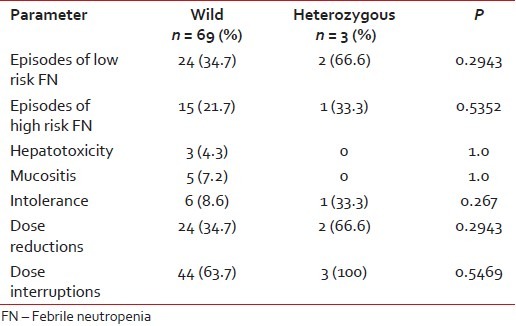
The number of episodes of low risk FN requiring oral antibiotics were 24/69 (34.7%) in wild type and 2/3 (66.6%) in heterozygous type (P = 0.2943). The number of episodes of high risk FN requiring admission and intravenous antibiotics in wild and heterozygous variant were 15/69 (21.7%) and 1/3 (33.3%), respectively (P = 0.5352). Grade 1 and 2 hepatotoxicity in wild variant was 3/69 (4.3%) and none in the heterozygous variant (P = 1.0). Grade 1 and 2 mucositis in wild variant was 5/69 (7.2%) and none in heterozygous variant (P = 1.0). There were no grade 3 or 4 hepatotoxicity and mucositis [Table 2].
Table 2
Comparison of toxicities between two groups
|
6-mercaptopurine dose was decreased by (25-75%) in 24/69 (34.7%) in wild variant and 2/3 (66.6%) in heterozygous variant (P = 0.2943). Dose interruptions, that is, children who were not on 6-MP for more than 2 weeks were 44/69 (63.7%) and 3/3 (100%) in wild and heterozygous variants, respectively (P = 0.5469).
Intolerance to 6-MP was 6/69 (8.6%) in wild variant and 1/3 (33.3%) in heterozygous variant (P = 0.267). Median daily dose of 6-MP was 42.92 mg/m2 /day (range, 11.4-56.8) in wild variant and 41.5 mg/m2 /day (range, 24.5-50.8) in heterozygous variant. Median weekly dose of MTX in mg/m2 /week was 9.36 mg/m2 (range, 4.5-11.2) in wild variant and 9.3 mg/m2 /week range (5.6-9.6) in heterozygous variant.
Median number of weeks taken to complete one cycle of oral maintenance in wild and heterozygous variant were 14.4 (range, 11.4-26.8) and 14.2 (range, 14.1-18), respectively.
6-mercaptopurine is a prodrug and it is activated to its active form by hypoxanthine phosphoribosyltransferase and inosine triphosphate pyrophosphatase and several others enzymes. TPMT catalyses 6-MP to its inactive form methylmercaptopurine nucleotides (6-MMPN). Genetic polymorphisms of the genes coding for these enzyme may influence the therapeutic and toxic effects of 6-MP.[14] Among these enzymes, the TPMT genotype, its enzyme activity and toxicity due to thiopurines is well-established.
Deficiency of TPMT may lead to increased accumulation of thioguanine nucleotides leading to hematopoietic toxicity. Conversely, increased 6-MMPNs production in patients with the wild type activity results in increased risk for hepatotoxicity.[16]
Homozygous variants have low enzyme activity leading to fatal toxicities. Heterozygous variants have intermediate enzyme activity; toxicity is moderate, requiring reduction in drug.
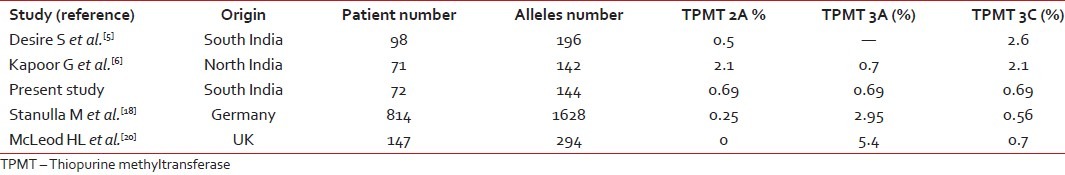
Racial differences in TPMT polymorphisms are well-established. Various studies reported 2-14% prevalence of TPMT polymorphisms among different population.[15,17,18] Higher incidence has been reported in Caucasians as compared to Asians[15,17,18] [Table 1].
Table 1
Frequency distribution of TPMT polymorphism in different population
|
In the present study, the incidence of heterozygous mutation was 4.2%. The frequency of heterozygous polymorphisms in one study from North India reported by Kapoor G et al. was 10% and none were homozygous.[6] In a study from South India by Desire S et al., 4 (4.4%) out of 98 children were heterozygous for 3C and 1 (1%) out of 98 patients was compound heterozygous for both 2A and 3C; none were homozygous for TPMT polymorphism.[5] Another study from North India in healthy population reported an incidence of 4% heterozygous polymorphisms of TPMT.[19] Kham SK et al. from Singapore, reported an incidence of 2.3% in multi ethnic migrant Asian population.[15] McLeod HL et al. reported the frequency of heterozygous variant as 10% and homozygous variant as 0.7% in British children with ALL.[20] Dokmanovic L et al. from Serbia and Montenegro reported frequency of heterozygous polymorphisms to be 10% and compound heterozygous to be 1% in children with ALL.[4] Frequency of TPMT polymorphisms in the present study was similar to South Indian and Asian studies and lower than the Caucasian population [Table 1].
The relative frequencies of heterozygous polymorphisms in the present study were 1/3 (33%) for 2A, 3A, 3C polymorphisms. Kapoor G et al. from North India reported that 3/7 (42.8%) were 3C and 3/7 (42.8%) were 2A and 1/7 (14.2%) were 3A.[6] In the South Indian study reported by Desire S et al., five patients 4 (80%) were heterozygous for 3C and one (20%) was compound heterozygous for both 2A and 3C.[5] Kham SK et al. reported the most common polymorphisms as 3C in multi ethnic population from South Asia.[15] McLeod HL et al. reported in British population an 3A as most common TPMT polymorphism.[20] Dokmanovic L et al. also reported as 3A as most common TPMT polymorphism.[4] 3C is the most common polymorphism in the present study and it is similar to that reported from the other Indian and Asian studies but different from to Caucasians where 3A is the most common.
Seventy-two had completed first oral maintenance cycle, and are evaluable for toxicity. Three out seventy-two were heterozygous and rest were wild for TPMT polymorphisms. Most common toxicity reported in our study was FN in first 3 months of therapy [Table 2].
Episodes of low risk FN requiring oral antibiotics in the present study were 24/69 (34.7%) in wild type children and 2/3 (66.6%) in heterozygous variant children. Episodes of high risk FN requiring intravenous antibiotics were 15/69 (21.7%) in wild type children and 1/3 (33.3%) in heterozygous variant children. Only 40% of FN episode are high risk needing intravenous antibiotics and rest were managed with oral antibiotics. Kapoor G et al. reported that in children treated for at least 1-year of maintenance (modified ALL-BFM 90 protocol), there were 46 episodes of FN in 64 children with wild allele and 5 episodes in seven children with mutant alleles.[6] Dokmanovic L et al. reported in children treated on modified ALL-BFM 90 protocol, in their retrospective group, in whom 6-MP was given without knowing the genotype. The FN episodes were more in children with the mutant allele. In the prospective group, as the dosage was adjusted according to the genotype, there was no episode of FN.[4] The dose of 6-MP in these both studies was 50 mg/m2 daily continuously for 2 years.
In the present study, 3 (4.2%) children on 6-MP developed grade 1-2 hepatic toxicity in wild allele group and none in heterozygous group. In the present study, 5 (7.2%) children had grade 1-2 oral mucositis in wild allele group and none in mutant allele group. Kapoor G et al. reported 14/64 (21.8%) children in wild allele group had mucositis and none in mutant allele group. In both studies, the incidence of mucositis was more in wild variant and less in mutant allele group.[6]
In the present study, intolerance to drug was noted in 6/69 (8.6%) children with wild allele group and 1/3 (33.3%) of children with heterozygous allele group. Kapoor G et al. reported vomiting in 17/64 children with wild allele group and 3/7 in heterozygous allele group.[6]
Dose reduction was required in 24/69 (34.7%) in children in wild allele group and 2/3 (66.6%) in children in heterozygous group in the present study. Desire S et al. in their study reported that 3/5 (60%) children in heterozygous group and 12/61 (20%) in children with wild allele required at least 10% dose reduction.[5]
Forty-four out of 69 children in the wild allele group required more than 2 weeks to recover from myelotoxicity, as were all the patients (3/3) in heterozygous group who required more than 2 weeks, recovering from toxicity in the present study. Dose interruption in our center could not be totally attributed to 6-MP toxicity. There are always other factors like socio economic status, distance to travel and natural calamities which may have profound impact on compliance in our population. In many studies, it is well-established that dose interruptions are more in heterozygous allele variants.[4,6,20]
Median number of weeks required to complete first oral maintenance cycle was 14.4 (range, 11.4-26.8 weeks) and median dose of 6-MP was 42.92 mg/day/m2 (range, 22.0-56.8 mg) and median dose of MTX was 9.36 mg/m2 /week (range, 4.5-11.2 mg) in children with wild allele.
Median number of weeks required to complete first oral maintenance cycle was 14.2 (range, 14.1-18 weeks) and median dose of 6-MP was 41.5 mg/day/m2 (range, 24.5-50.8 mg) and median dose of MTX was 9.3 mg/m2 /week (range, 5.6-9.6 mg) in children with heterozygous allele. Kapoor G et al. reported that the median dose of both 6-MP and MTX were lower in heterozygous group.[6]
We acknowledge the limitations of this study. This study is in a small cohort of 72 children and toxicity is reported in only the first cycle of maintenance. In the maintenance phase, MTX is given weekly and its influence on toxicity profile of 6-MP cannot be ruled out. Other confounding factors such as poor hygiene, lower socioeconomic status, quality of drugs, poor nutrition, and inter-current infections also could not be ruled out.
CONCLUSION
The frequency of TPMT polymorphisms in ALL children treated at our center is 4.2%. The most common heterozygous variants are 2, 3A, 3C.
ACKNOWLEDGMENTS
All the supporting departments at NIMS, Dr. Sudha Sinha, Dr. Satya Dattatreya Palanki and Dr. Ramana Dandamudi, Acton Biotech, Pune.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- Arora RS, Eden TO, Kapoor G. Epidemiology of childhood cancer in India. Indian J Cancer 2009;46:264-73.
- Yang JJ, Bhojwani D. Thiopurine S-methyltransferase pharmacogenetics in childhood acute lymphoblastic leukemia. Methods Mol Biol 2013;999:273-84.
- Appell ML, Wennerstrand P, Peterson C, Hertervig E, Mårtensson LG. Characterization of a novel sequence variant, TPMTFNx0128, in the human thiopurine methyltransferase gene. Pharmacogenet Genomics 2010;20:700-7.
- Dokmanovic L, Urosevic J, Janic D, Jovanovic N, Petrucev B, Tosic N, et al. Analysis of thiopurine S-methyltransferase polymorphism in the population of Serbia and Montenegro and mercaptopurine therapy tolerance in childhood acute lymphoblastic leukemia. Ther Drug Monit 2006;28:800-6.
- Desire S, Balasubramanian P, Bajel A, George B, Viswabandya A, Mathews V, et al. Frequency of TPMT alleles in Indian patients with acute lymphatic leukemia and effect on the dose of 6-mercaptopurine. Med Oncol 2010;27:1046-9.
- Kapoor G, Sinha R, Naithani R, Chandgothia M. Thiopurine S-methyltransferase gene polymorphism and 6-mercaptopurine dose intensity in Indian children with acute lymphoblastic leukemia. Leuk Res 2010;34:1023-6.
- Dorababu P, Naushad SM, Linga VG, Gundeti S, Nagesh N, Kutala VK, et al. Genetic variants of thiopurine and folate metabolic pathways determine 6-MP-mediated hematological toxicity in childhood ALL. Pharmacogenomics 2012;13:1001-8.
- Dorababu P, Nagesh N, Linga VG, Gundeti S, Kutala VK, Reddanna P, et al. Epistatic interactions between thiopurine methyltransferase (TPMT) and inosine triphosphate pyrophosphatase (ITPA) variations determine 6-mercaptopurine toxicity in Indian children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012;68:379-87.
- Iyer SN, Tilak AV, Mukherjee MS, Singhal RS. Genotype frequencies of drug-metabolizing enzymes responsible for purine and pyrimidine antagonists in a healthy Asian-Indian population. Biochem Genet 2012;50:684-93.
- Umamaheswaran G, Krishna Kumar D, Kayathiri D, Rajan S, Shewade DG, Dkhar SA, et al. Inter and intra-ethnic differences in the distribution of the molecular variants of TPMT, UGT1A1 and MDR1 genes in the South Indian population. Mol Biol Rep 2012;39:6343-51.
- Ameyaw MM, Collie-Duguid ES, Powrie RH, Ofori-Adjei D, McLeod HL. Thiopurine methyltransferase alleles in British and Ghanaian populations. Hum Mol Genet 1999;8:367-70.
- Yates CR, Krynetski EY, Loennechen T, Fessing MY, Tai HL, Pui CH, et al. Molecular diagnosis of thiopurine S-methyltransferase deficiency: Genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med 1997;126:608-14.
- Advani S, Pai S, Venzon D, Adde M, Kurkure PK, Nair CN, et al. Acute lymphoblastic leukemia in India: an analysis of prognostic factors using a single treatment regimen. Ann Oncol 1999;10:167-76.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006;24:715-29.
- Kham SK, Tan PL, Tay AH, Heng CK, Yeoh AE, Quah TC. Thiopurine methyltransferase polymorphisms in a multiracial Asian population and children with acute lymphoblastic leukemia. J Pediatr Hematol Oncol 2002;24:353-9.
- Dervieux T, Meshkin B, Neri B. Pharmacogenetic testing: Proofs of principle and pharmacoeconomic implications. Mutat Res 2005;573:180-94.
- McLeod HL, Krynetski EY, Relling MV, Evans WE. Genetic polymorphism of thiopurine methyltransferase and its clinical relevance for childhood acute lymphoblastic leukemia. Leukemia 2000;14:567-72.
- Stanulla M, Schaeffeler E, Möricke A, Coulthard SA, Cario G, Schrauder A, et al. Thiopurine methyltransferase genetics is not a major risk factor for secondary malignant neoplasms after treatment of childhood acute lymphoblastic leukemia on Berlin-Frankfurt-Münster protocols. Blood 2009;114:1314-8.
- Kapoor G, Maitra A, Somlata, Brahmachari V. Application of SNaPshot for analysis of thiopurine methyltransferase gene polymorphism. Indian J Med Res 2009;129:500-5.
- McLeod HL, Coulthard S, Thomas AE, Pritchard SC, King DJ, Richards SM, et al. Analysis of thiopurine methyltransferase variant alleles in childhood acute lymphoblastic leukaemia. Br J Haematol 1999;105:696-
References
- Arora RS, Eden TO, Kapoor G. Epidemiology of childhood cancer in India. Indian J Cancer 2009;46:264-73.
- Yang JJ, Bhojwani D. Thiopurine S-methyltransferase pharmacogenetics in childhood acute lymphoblastic leukemia. Methods Mol Biol 2013;999:273-84.
- Appell ML, Wennerstrand P, Peterson C, Hertervig E, Mårtensson LG. Characterization of a novel sequence variant, TPMTFNx0128, in the human thiopurine methyltransferase gene. Pharmacogenet Genomics 2010;20:700-7.
- Dokmanovic L, Urosevic J, Janic D, Jovanovic N, Petrucev B, Tosic N, et al. Analysis of thiopurine S-methyltransferase polymorphism in the population of Serbia and Montenegro and mercaptopurine therapy tolerance in childhood acute lymphoblastic leukemia. Ther Drug Monit 2006;28:800-6.
- Desire S, Balasubramanian P, Bajel A, George B, Viswabandya A, Mathews V, et al. Frequency of TPMT alleles in Indian patients with acute lymphatic leukemia and effect on the dose of 6-mercaptopurine. Med Oncol 2010;27:1046-9.
- Kapoor G, Sinha R, Naithani R, Chandgothia M. Thiopurine S-methyltransferase gene polymorphism and 6-mercaptopurine dose intensity in Indian children with acute lymphoblastic leukemia. Leuk Res 2010;34:1023-6.
- Dorababu P, Naushad SM, Linga VG, Gundeti S, Nagesh N, Kutala VK, et al. Genetic variants of thiopurine and folate metabolic pathways determine 6-MP-mediated hematological toxicity in childhood ALL. Pharmacogenomics 2012;13:1001-8.
- Dorababu P, Nagesh N, Linga VG, Gundeti S, Kutala VK, Reddanna P, et al. Epistatic interactions between thiopurine methyltransferase (TPMT) and inosine triphosphate pyrophosphatase (ITPA) variations determine 6-mercaptopurine toxicity in Indian children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012;68:379-87.
- Iyer SN, Tilak AV, Mukherjee MS, Singhal RS. Genotype frequencies of drug-metabolizing enzymes responsible for purine and pyrimidine antagonists in a healthy Asian-Indian population. Biochem Genet 2012;50:684-93.
- Umamaheswaran G, Krishna Kumar D, Kayathiri D, Rajan S, Shewade DG, Dkhar SA, et al. Inter and intra-ethnic differences in the distribution of the molecular variants of TPMT, UGT1A1 and MDR1 genes in the South Indian population. Mol Biol Rep 2012;39:6343-51.
- Ameyaw MM, Collie-Duguid ES, Powrie RH, Ofori-Adjei D, McLeod HL. Thiopurine methyltransferase alleles in British and Ghanaian populations. Hum Mol Genet 1999;8:367-70.
- Yates CR, Krynetski EY, Loennechen T, Fessing MY, Tai HL, Pui CH, et al. Molecular diagnosis of thiopurine S-methyltransferase deficiency: Genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med 1997;126:608-14.
- Advani S, Pai S, Venzon D, Adde M, Kurkure PK, Nair CN, et al. Acute lymphoblastic leukemia in India: an analysis of prognostic factors using a single treatment regimen. Ann Oncol 1999;10:167-76.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006;24:715-29.
- Kham SK, Tan PL, Tay AH, Heng CK, Yeoh AE, Quah TC. Thiopurine methyltransferase polymorphisms in a multiracial Asian population and children with acute lymphoblastic leukemia. J Pediatr Hematol Oncol 2002;24:353-9.
- Dervieux T, Meshkin B, Neri B. Pharmacogenetic testing: Proofs of principle and pharmacoeconomic implications. Mutat Res 2005;573:180-94.
- McLeod HL, Krynetski EY, Relling MV, Evans WE. Genetic polymorphism of thiopurine methyltransferase and its clinical relevance for childhood acute lymphoblastic leukemia. Leukemia 2000;14:567-72.
- Stanulla M, Schaeffeler E, Möricke A, Coulthard SA, Cario G, Schrauder A, et al. Thiopurine methyltransferase genetics is not a major risk factor for secondary malignant neoplasms after treatment of childhood acute lymphoblastic leukemia on Berlin-Frankfurt-Münster protocols. Blood 2009;114:1314-8.
- Kapoor G, Maitra A, Somlata, Brahmachari V. Application of SNaPshot for analysis of thiopurine methyltransferase gene polymorphism. Indian J Med Res 2009;129:500-5.
- McLeod HL, Coulthard S, Thomas AE, Pritchard SC, King DJ, Richards SM, et al. Analysis of thiopurine methyltransferase variant alleles in childhood acute lymphoblastic leukaemia. Br J Haematol 1999;105:696-