 PDF
PDF  Views
Views  Share
Share


A Rare Extramedullary and Extralymphoid Presentation of Mixed Phenotypic Blastic Hematolymphoid Neoplasm: A Study of Two Cases
CC BY-NC-ND 4.0 · Indian J Med Paediatr Oncol 2017; 38(03): 394-397
DOI: DOI: 10.4103/ijmpo.ijmpo_94_16
Abstract
Mixed phenotype acute leukemia (MPAL) is a rare hematolymphoid neoplasm, representing only 3%–5% of acute leukemia. Although MPAL has been sufficiently described in the literature, its extramedullary presentation as a solitary lesion without leukemic (bone marrow [BM]) involvement is rarely described. We are presenting two cases of mixed phenotypic blastic hematolymphoid neoplasms without leukemic involvement at disease presentation in 8-year-old female and 21-year-old male patients. Both the cases had extralymphatic bone involvement in the form of solitary bone lesion. Initially, there was no leukemic involvement in both the cases, but the second case progressed to acute leukemia during the course of the disease. On immunophenotypic evaluation, both the cases revealed blasts showing unequivocal evidence of myeloid and B-lymphoid lineage commitment. These cases were difficult to categorize either into MPAL as the BM was not involved or into lymphoblastic lymphoma due to coexpression of myeloid differentiation. Therefore, we chose to classify them as a bi/mixed phenotypic blastic hematolymphoid neoplasm. Detailed immunophenotypic analysis either by immunohistochemistry or flow cytometric immunophenotyping is important for the diagnosis of such cases as they have a poor prognosis.
Publication History
Article published online:
04 July 2021
© 2017. Indian Society of Medical and Paediatric Oncology. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/.)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
Mixed phenotype acute leukemia (MPAL) is a rare hematolymphoid neoplasm, representing only 3%–5% of acute leukemia. Although MPAL has been sufficiently described in the literature, its extramedullary presentation as a solitary lesion without leukemic (bone marrow [BM]) involvement is rarely described. We are presenting two cases of mixed phenotypic blastic hematolymphoid neoplasms without leukemic involvement at disease presentation in 8-year-old female and 21-year-old male patients. Both the cases had extralymphatic bone involvement in the form of solitary bone lesion. Initially, there was no leukemic involvement in both the cases, but the second case progressed to acute leukemia during the course of the disease. On immunophenotypic evaluation, both the cases revealed blasts showing unequivocal evidence of myeloid and B-lymphoid lineage commitment. These cases were difficult to categorize either into MPAL as the BM was not involved or into lymphoblastic lymphoma due to coexpression of myeloid differentiation. Therefore, we chose to classify them as a bi/mixed phenotypic blastic hematolymphoid neoplasm. Detailed immunophenotypic analysis either by immunohistochemistry or flow cytometric immunophenotyping is important for the diagnosis of such cases as they have a poor prognosis.
Background
Mixed phenotype acute leukemia (MPAL) is a rare disease, representing only 3%–5% of acute leukemia of all age groups.[1] MPAL comprises either single blasts population with the presence of more than one lineage-specific markers (formerly called as biphenotypic leukemia) or a presence of two distinct blast populations with two distinct lineage-specific markers (formerly called as bilineage leukemia).[1] The World Health Organisation (WHO) definition of MPAL is based on the expression of lineage-specific markers of two or more lineages that include surface/cytoplasmic CD3 for T-lymphoid, myeloperoxidase (MPO) for myeloid, expression of any two of CD14, CD64, CD11c, and lysozyme, or cytochemistry (esterase tests) for monocytic lineage.[1] For B cell lineage assignment, the WHO has given two criteria, that is, either the strong expression of CD19 together with another B cell-associated marker or in cases with weak CD19, the expression of at least two B-lineage markers.[1] In addition, the WHO recognizes two distinct categories, MPAL with the t(9;22)(q34;q11)/BCR-ABL1 and MPAL with t(v; 11q23)/mixed lineage leukemia (MLL) rearrangement. The remaining cases are designated as MPAL not otherwise specified. Although MPAL has been described in the literature, presentation as a solitary lesion without leukemic (bone marrow [BM]) involvement is rarely described. Identification of such cases using adequate immunophenotypic markers is very important as these cases had a poor prognosis, required intensive chemotherapy, and are resistant to treatment with a poor outcome.[1] We are presenting two rare cases of mixed phenotypic blastic hematolymphoid neoplasms without leukemic involvement at disease presentation highlighting the importance of adequate immunophenotypic workup for the diagnosis of such lesions.
Case Reports
Case 1
An 8-year-old female presented with the right cheek swelling for 2 months with no additional symptoms. The swelling was ill-defined, firm, and nontender. Her general and systemic examinations were normal. Positron emission tomography (PET) scan with F-18 fludeoxyglucose (FDG) revealed an FDG-avid soft tissue mass measuring 3.6 cm × 3.5 cm with maximum standardized uptake value (SUV) of 11.88 in the right maxillary sinus with complete opacification and erosion of the adjacent superior alveolus with thinning of the walls of the maxillary sinus with an extramaxillary soft tissue component. Hematology values were as follows: hemoglobin (Hb) concentration – 13.1 g/dl; white blood cell (WBC) count – 7.8 × 109/L; platelets – 400 × 109/L; and peripheral blood smear (PBS) examination did not reveal any abnormal cells or blasts. Subsequent BM examination revealed normocellular to mildly hypocellular BM with trilineage hematopoiesis. No blasts or abnormal cells were identified. Cytology of cerebrospinal fluid also did not reveal any abnormal cells.
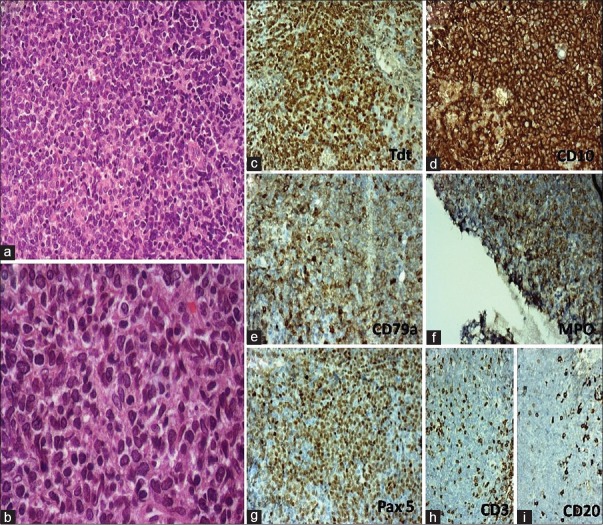
The right maxillary sinus biopsy showed diffuse proliferation of medium size cells with scant cytoplasm and blastic chromatin [Figure 1]. Mild pleomorphism in the size of the tumor cells with high mitotic activity was noted. On immunohistochemistry (IHC) [Figure 1], the tumor cells were positive for CD99 (Mic2), diffusely positive for B cell markers such as CD10, CD79a, PAX5 and also expressed MPO and weakly CD45 (LCA) but negative for CD20, CD3, CD5, desmin, and chromogranin. In addition, nearly all tumor cells showed nuclear positivity for terminal deoxynucleotidyl transferase (TdT). Fluorescence in situ hybridization on interphase cells of formalin-fixed paraffin-embedded tissue revealed negative results for BCR/ABL using locus-specific identifier (LSI) BCR/ABL dual fusion translocation probe and for MLL using LSI MLL (11q23) break apart rearrangement probe; however, nearly 40% of cells revealed trisomy of chromosome 22. Thus, histological and IHC findings were consistent with bi/mixed phenotypic blastic hematolymphoid neoplasm with B-lymphoblastic lymphoma with coexpression of the myeloid lineage-specific marker and MPO.
| Figure 1:(a and b) A bulky ulcerative cauliflower-like diffuse anterior neck mass measuring 8 cm × 8 cm involving the submental and anterior cervical region and bilateral axillary lymphadenopathy measuring 3 cm × 2 cm each. (c) Sparse large atypical lymphoid cells over a background of necrosis and acute inflammatory cells. (d) Atypical lymphoid cells some amount of eosinophilic cytoplasm, fine chromatin and prominent central nucleoli scattered over a loose fibro collagenous stroma. Plasmacytoid morphology
She was started on treatment with institutional acute lymphoblastic leukemia protocol called MCP-841 with standard 4-drug induction followed by a consolidation block with intermediate-dose cytarabine known to be active in myeloid neoplasms, interim maintenance, and delayed intensification. After the end of induction, she had a good response in maxillary mass with complete metabolic and a partial morphological response with calcification of mass. She was continued on postinduction chemotherapy and received consolidative radiotherapy to the right maxilla in a dose of 25.2 grays in 14 fractions during interim maintenance. Her postradiation contrast-enhanced computed tomography scan revealed only thickening and sclerosis of the wall of maxillary sinus suggestive of healing. She is currently continuing on oral maintenance chemotherapy and is doing well after 3 maintenance cycles at 18-month follow-up.
Case 2
A 21-year-old male was being investigated for an acute onset of pain involving the left hip for 6–8 weeks. The magnetic resonance imaging revealed a permeative lesion involving the head and upper shaft of the femur. The needle core biopsy from the lesion was performed in another hospital which was reported as suspicious of a primitive neuroectodermal tumor and was referred to our institute. His laboratory workup in our hospital revealed normal complete blood count with Hb concentration – 14.2 g/dl; WBC count – 5.7 × 109/L; platelets – 278 × 109/L; and PBS did not reveal any abnormal cells or blasts. The serum lactate dehydrogenase was mildly raised to 216 U/L. PET scan with F-18 FDG revealed hypermetabolic disease involving the head, neck, and proximal one-third shaft of the left femur with maximum SUV of 7.6 with no obvious cortical destruction or soft tissue mass. In addition, the proximal end of the left fibula also showed an active disease with maximum SUV of 3.59. There was no evidence of active disease elsewhere in the body.
A biopsy of the lesion was reviewed which showed sheets of medium-sized tumor cells with scant cytoplasm and blastic chromatin with few crushing artifacts. Mild pleomorphism of tumor cells with focal apoptosis was noted. These features were consistent with a high-grade blastic neoplasm. On IHC, the tumor cells showed diffuse membranous expression of MIC2, whereas TdT showed weak, but distinct nuclear reactivity. In B-lymphoid lineage markers, CD10 and CD20 were diffusely positive while PAX5 was negative. In addition, blasts expressed MPO; however, CD117 did not reveal any positive expression.
The BM study was omitted initially as there was no evidence of any other sites or marrow involvement or uptake by PET scan. However, BM was done after a month of diagnosis, and it revealed 54% of blasts which were negative for cytochemical MPO. We performed eight-color flow cytometric immunophenotyping (FCI) using a comprehensive antibody panel [Table 1]. Cells were acquired on Navios (Beckman Coulter [BC]) flow cytometer using Navios software, and data were analyzed using Kaluza software (BC).
Table 1
Comprehensive antibody panel

| Immunophenotypic analysis [Figure 2] showed approximately 43% of viable cells were abnormal blasts. The abnormal blasts revealed a B-lymphoid lineage origin expressing CD10, CD19, CD20, and CD22 but negative for cytoplasmic CD79a. It also showed expression of CD34 and human leukocyte antigen–antigen D. In addition, they showed cytoplasmic MPO expression but were negative for rest of the other markers tested. BM trephine biopsy also showed a sheet of blasts expressing CD10, CD20, and MPO. Cytogenetics revealed no evidence of BCR/ABL fusion: t(9;22), AML1/ETO: t(8;21), and MLL rearrangement.

| Figure 2:Magnetic resonance imaging T1-weighted coronal (a), T2-weighted coronal (b), T2-weighted axial (c), and STIR axial (d) images showing a well-defined round to oval large mass (arrow) in deep subcutaneous and intermuscular compartment of left posterolateral abdominal wall. The lesion measures 9.7 cm × 7.5 cm in transverse and anteroposterior dimension, respectively. The lesion is heterogeneously hyperintense on T1 weighted and T2 weighted as compared to muscles. There is no signal drop on fat suppressed sequence suggestive of absent fatty component. The central part of lesion shows T2 hyperintense areas suggestive of necrosis. There is no extension into spinal canal. Chest X-ray posteroanterior view, (e) left-sided pleural effusion
All features were consistent with bi/mixed phenotypic blastic hematolymphoid neoplasm as B-lymphoblastic lymphoma with coexpression of the myeloid marker and MPO that progressed to mixed phenotypic acute leukemia (B/myeloid leukemia) during a course of time.
Discussion
Most of acute leukemias are classified into myeloid, B-lymphoid, or T-lymphoid origin according to the antigenic expression of the blasts. However, in some cases, it is difficult to categorize them into specific lineage because of the expression of both lymphoid and myeloid lineage-specific antigens in the blast cells, and these leukemias have been designated as MPAL in the WHO.[1] Although MPAL has been described in the literature, its presentation as a solitary lesion without leukemic involvement at the time of diagnosis is rarely described. To the best of our knowledge, only ten cases have been published in the literature,[2,3,4] and ours is the first report of two cases from India. Largest case series of six cases of biphenotypic lymphoma (B and T) showed hypercellular BM with myeloid preponderance and eosinophilia with the t(8,13) association.[2] One case had recurrent episodes of unexplained cervical lymphadenopathy over a period of several years and an acute onset of myasthenia gravis 1 year before patient's presentation with worsening respiratory difficulty,[3] and another case had nontender neck lymph nodes for 9 months and was being evaluated for lymphoma.[4] In these cases, tumor cells expressed both myeloid and T-lymphoid markers and lymph nodes being the most common site.
In the present study, both cases had extralymphatic bony involvement with no leukemic involvement. Notably, involvement in the second case, initially there was no leukemic involvement but progressed to leukemia during the course of the disease. Both cases had blasts showing unequivocal evidence of myeloid and B-lymphoid commitment as shown by the expression of lineage-specific antigens. Both cases could be diagnosed due to comprehensive immunophenotyping on biopsy. If we would have been restricted our IHC to only lymphoid panel, these would have been diagnosed as B-lymphoblastic lymphoma. Both cases were difficult to categorize either into MPAL as the BM was not involved or into lymphoblastic lymphoma because of coexpression of myeloid differentiation. Therefore, we chose to classify them as a bi/mixed phenotypic blastic hematolymphoid neoplasm.
Conclusion
MPAL presenting as an extramedullary and extralymphatic lesion is extremely rare and difficult to categorize. Detailed immunophenotypic analysis on IHC or FCI is necessary for the diagnosis of such cases, and it is important to diagnose and classify them correctly as they harbor a poor prognosis.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Swerdlow S, Campo E, Harris N, Jaffe E, Pileri S, Stein H, et al. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008. p. 150-5.
- Vega F, Medeiros LJ, Davuluri R, Cromwell CC, Alkan S, Abruzzo LV. t(8;13)-positive bilineal lymphomas: Report of 6 cases. Am J Surg Pathol 2008;32:14-20.
- Childs CC, Chrystal GS, Strauchen JA. Biphenotypic lymphoblastic lymphoma. An unusual tumor with lymphocytic and granulocytic differentiation. Cancer 1986;57:1019-23.
- Bayraktar UD, Barker J, Pereira D, Glick DZ, Byrne GE Jr., Escalón MP. An unusual presentation of extramedullary biphenotypic lymphoblastic lymphoma. Am J Hematol 2009;84:616-7.
| Figure 1:(a and b) A bulky ulcerative cauliflower-like diffuse anterior neck mass measuring 8 cm × 8 cm involving the submental and anterior cervical region and bilateral axillary lymphadenopathy measuring 3 cm × 2 cm each. (c) Sparse large atypical lymphoid cells over a background of necrosis and acute inflammatory cells. (d) Atypical lymphoid cells some amount of eosinophilic cytoplasm, fine chromatin and prominent central nucleoli scattered over a loose fibro collagenous stroma. Plasmacytoid morphology
| Figure 2:Magnetic resonance imaging T1-weighted coronal (a), T2-weighted coronal (b), T2-weighted axial (c), and STIR axial (d) images showing a well-defined round to oval large mass (arrow) in deep subcutaneous and intermuscular compartment of left posterolateral abdominal wall. The lesion measures 9.7 cm × 7.5 cm in transverse and anteroposterior dimension, respectively. The lesion is heterogeneously hyperintense on T1 weighted and T2 weighted as compared to muscles. There is no signal drop on fat suppressed sequence suggestive of absent fatty component. The central part of lesion shows T2 hyperintense areas suggestive of necrosis. There is no extension into spinal canal. Chest X-ray posteroanterior view, (e) left-sided pleural effusion
References
- Swerdlow S, Campo E, Harris N, Jaffe E, Pileri S, Stein H, et al. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008. p. 150-5.
- Vega F, Medeiros LJ, Davuluri R, Cromwell CC, Alkan S, Abruzzo LV. t(8;13)-positive bilineal lymphomas: Report of 6 cases. Am J Surg Pathol 2008;32:14-20.
- Childs CC, Chrystal GS, Strauchen JA. Biphenotypic lymphoblastic lymphoma. An unusual tumor with lymphocytic and granulocytic differentiation. Cancer 1986;57:1019-23.
- Bayraktar UD, Barker J, Pereira D, Glick DZ, Byrne GE Jr., Escalón MP. An unusual presentation of extramedullary biphenotypic lymphoblastic lymphoma. Am J Hematol 2009;84:616-7.