 PDF
PDF  Views
Views  Share
Share


Role of Cytogenetics and Fluorescence In Situ Hybridization in the Laboratory Workup of Acute Myeloid Leukemias
CC BY 4.0 · Indian J Med Paediatr Oncol 2023; 44(06): 543-553
DOI: DOI: 10.1055/s-0043-1768052

Abstract
A new understanding of acute myeloid leukemia as a varied group of unique biologic entity has emerged, as a result of the identification of various chromosomal aberrations and their association with clinical prognosis and diagnosis. Following induction treatment, cytogenetic examination can establish the presence of any residual malignant cells, it's recurrence, clonal evolution if any, or the formation of novel abnormalities. The G-banded karyotype has been the gold standard method for detecting all of these aberrations for years. The capacity to examine the entire genome through karyotype analysis quickly enabled the detection of deletions, duplications, and structural rearrangements across every chromosome, and the more frequent ones were associated with particular aberrant clinical symptoms. Fluorescence in situ hybridization (FISH) is a sensitive technology that aids in differential diagnosis or therapeutic planning and provides rapid results. Furthermore, the combination of cytogenetic and molecular profiling enables a more precise evaluation of disease prognosis, diagnosis, classification, risk stratification, and patient treatment. Interphase FISH analysis, in conjunction with G-banded chromosomal analysis, can be used as a major testing tool for the evaluation of hematological neoplasms. For accurate and consistent descriptions of genomic changes identified by karyotyping and FISH, a specified terminology is necessary. The International System for Human Cytogenomic Nomenclature is the main source and provides instructions for documenting cytogenetic and molecular findings in laboratory reports. This review discusses the two methods, karyotyping and FISH, their advantages and limitations, sample requirements, various FISH probes that are used, nomenclature for results reporting, and the necessary quality control measures.
Keywords
WHO classification - prognosis - FISH - cytogenetics - amplification - probesAuthors' Contributions
H.J. was responsible for concept, design, definition of intellectual content, literature search, and manuscript preparation. D.S. was responsible for manuscript editing and manuscript review. The manuscript has been read and approved by all the authors, and the requirements for authorship have been met and each author believes that the manuscript represents honest work.
Supplementary MaterialPublication History
Article published online:
27 November 2023
© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
Abstract
A new understanding of acute myeloid leukemia as a varied group of unique biologic entity has emerged, as a result of the identification of various chromosomal aberrations and their association with clinical prognosis and diagnosis. Following induction treatment, cytogenetic examination can establish the presence of any residual malignant cells, it's recurrence, clonal evolution if any, or the formation of novel abnormalities. The G-banded karyotype has been the gold standard method for detecting all of these aberrations for years. The capacity to examine the entire genome through karyotype analysis quickly enabled the detection of deletions, duplications, and structural rearrangements across every chromosome, and the more frequent ones were associated with particular aberrant clinical symptoms. Fluorescence in situ hybridization (FISH) is a sensitive technology that aids in differential diagnosis or therapeutic planning and provides rapid results. Furthermore, the combination of cytogenetic and molecular profiling enables a more precise evaluation of disease prognosis, diagnosis, classification, risk stratification, and patient treatment. Interphase FISH analysis, in conjunction with G-banded chromosomal analysis, can be used as a major testing tool for the evaluation of hematological neoplasms. For accurate and consistent descriptions of genomic changes identified by karyotyping and FISH, a specified terminology is necessary. The International System for Human Cytogenomic Nomenclature is the main source and provides instructions for documenting cytogenetic and molecular findings in laboratory reports. This review discusses the two methods, karyotyping and FISH, their advantages and limitations, sample requirements, various FISH probes that are used, nomenclature for results reporting, and the necessary quality control measures.
Keywords
WHO classification - prognosis - FISH - cytogenetics - amplification - probesIntroduction
Acute myeloid leukemia (AML) is the most common adult leukemia and is characterized by clonal expansion of immature blast cells in the peripheral blood and bone marrow and is genetically heterogeneous with variable prognosis.[1] AML predominately affects those over 60 years of age, with progressively dismal prognosis in advanced age, but can develop in children and young adults too.[2] [3] Acquired clonal chromosome aberrations can be seen in the majority of AML patients. The most important initiating step in a significant proportion of adult and pediatric AML is the generation of chimeric fusions arising due to events such as balanced translocations and/or inversions/insertions in hematopoietic stem cells which have been identified as recurrent genetic abnormalities by the World Health Organization (WHO) classification 2008.[4] These recurrent genetic abnormalities are sufficient for diagnosing AML regardless of blast count in bone marrow and also paved the way for molecular studies that identified genes involved in the process of leukemogenesis. The discovery of specific chromosomal abnormalities and their relationship to cytomorphologic features, immunophenotype, and clinical outcome has led to a new understanding of AML as a diverse group of distinct biologic entities. The clinical significance of cytogenetic findings in AML for classification and understanding of pathogenetic mechanisms is growing, as evidenced by the WHO classification of AML.[5]
Implications of chromosomal aberrations in the determination of clinical outcome and its value as an independent prognostic indicator were established by studies in large series of AML patients.[6] [7] [8] Based on chromosomal abnormalities and gene mutations, guidelines and risk scoring systems have been developed by the National Comprehensive Cancer Network and the European Leukemia Net to help physicians design tailored therapeutic strategies. The disease can be categorized into favorable, intermediate, or adverse-risk groups ([Table 1]). For example, t(15;17)(q24.1;q21), [PML::RARA], t(8;21)(q22;q22) [RUNX1::RUNX1T1], and inv(16)(p13.1q22) [CBFB::MYH11] are associated with a favorable outcome, whereas inv(3)(q21q26.2) or t(3;3)(q21;q26.2) [RPN1::MECOM], t(6;9)(p23;q34) [DEK::NUP214], and KMT2A (11q23) rearrangements fall under adverse risk and are associated with a dismal prognosis.[9] [10] Cytogenetic testing is critical in the selection of targeted therapy for various leukemias. In acute promyelocytic leukemia (APL), novel fusion protein PML::RARA is generated by a translocation between chromosomes 15 and 17 and is effectively treated with targeted therapy, ATRA (all-trans retinoic acid).[11] [12] Cytogenetic analysis can confirm the residual disease, relapse, clonal evolution, or the emergence of new anomalies after induction chemotherapy.[13] [14]
|
Risk category |
Cytogenetic abnormality |
|---|---|
|
Favorable |
RUNX1::RUNX1T1-t(8;21)(q22;q22) CBFB::MYH11-inv(16)(p13.1q22)/t(16;16)(p13.1;q22) |
|
Intermediate |
MLLT3::KMT2A-t(9;11)(p21.3;q23.3) Cytogenetic abnormalities not categorized as favorable or adverse |
|
Adverse |
DEK::NUP214-t(6;9)(p23;q34.1) |
|
KMT2A rearrangements-t(v;11)(?;q23.3) |
|
|
BCR::ABL1-t(9;22)(q34.1;q11.2) |
|
|
GATA2::MECOM- inv(3)(q21.3q26.2)/t(3;3)(q21.3;q26.2) |
|
|
MECOM::?-t(3q26.2;?) |
|
|
KAT6A::CREBBP-t(8;16)(p11;p13) |
|
|
Monosomy 5/del(5q), Monosomy 7, Monosomy 17/abn(17p) |
|
|
Complex karyotypes, Monosomal karyotypes |
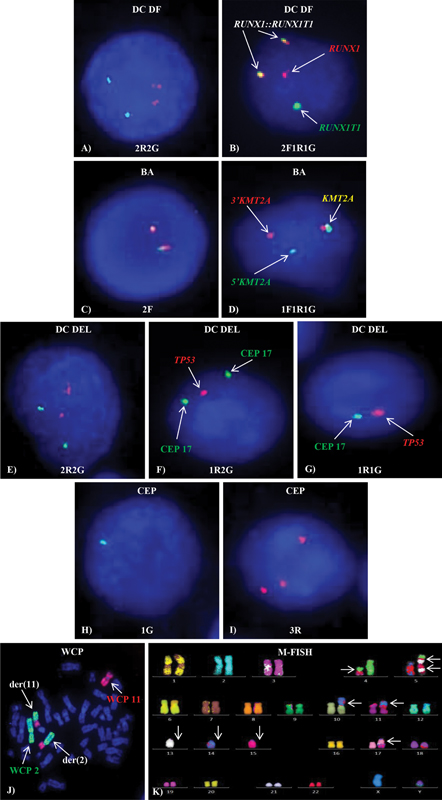
Figure 1:Different types of FISH probes. Normal (A) 2R2G, (C) 2F, (E) 2R2G and abnormal (B) 2F1R1G, (D) 1F1R1G, (F) 1R2G, (G) 1R1G, (H) 1G, (I) 3R FISH signal patterns on interphase cells (J) WCP showing t(2;11) and (K) M-FISH showing complex karyotype on metaphase.
Reporting of Conventional Cytogenetics and Fluorescence In Situ Hybridization Results
A defined nomenclature is essential for the correct and uniform description of genomic alterations discovered by karyotyping and FISH. The ISCN is the primary resource and offers guidelines for describing cytogenetic and molecular findings in laboratory reports. These laboratory reports serve as records that must be understandable, accurate, and should provide all the details necessary for the correct interpretation of the cytogenetic findings to the clinicians. The chromosome aberrations are denoted by a series of symbols and short terminologies. For example, a reciprocal exchange of chromosome segments between two different chromosomes is defined as translocation and is abbreviated as “t.” A karyotype is described as the total number of chromosomes followed by a sex chromosome complement separated by comma, a set of parentheses describing the chromosomes, their bands, and subbands involved in the aberration separated by a semicolon. A male patient with 46 chromosomes with a translocation between chromosomes 9 and 22 is described as 46,XY,t(9;22)(q34.1;q11.2), while a normal male karyotype is described as 46,XY.
Similarly, interphase FISH results begin with “nuc ish” which stands for nuclear in situ hybridization, followed by locus designation in parentheses, a multiplication sign (X) and the number of signals seen. If the number of signals for each probe is the same, the multiplication sign “X” is outside the parentheses. If the number of hybridization signals varies, then the multiplication sign “X” is inside the parentheses and the number of cells scored is placed in square brackets “[ ].” The nomenclature of FISH depends on the type of FISH probe used. As described in the earlier section, for a BCR/ABL1 DC DF probe, a normal cell with two red and two green signals is described as nuc ish(ABL1,BCR)X2[200], while an abnormal interphase cell with a reciprocal fusion between two differentially labeled genes, ABL1 and BCR, produces 2F1R1G signal pattern which is described as nuc ish(ABL1,BCR)X3(ABL1 con BCRX2) [196/200] where “con” stands for connected.[24]
Conventional Cytogenetics and Fluorescence In Situ Hybridization Strategy in Acute Myeloid Leukemia
In a cytogenetic laboratory, conventional karyotyping and selection of FISH probes targeting specific recurring abnormalities in AML are based on their diagnostic and prognostic utility ([Fig. 2], [Table 2]). Both, CC and FISH should be concurrently applied for workup of newly diagnosed and follow-up AML cases. As FISH has shorter turnaround time than CC, prognostically relevant AML-associated cytogenetic aberrations, cryptic rearrangements, and low-level abnormalities can be reported within 2 to 3 days aiding in therapeutic decisions. At the same time, in the absence of recurrent cytogenetic aberrations, CC helps in the detection of other abnormalities for which FISH probes are not available.
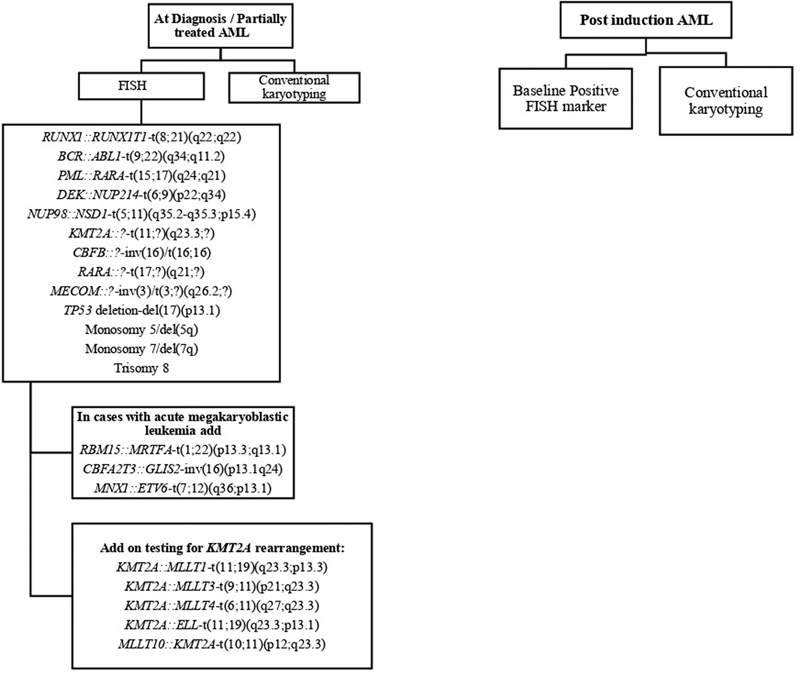
| Figure 2:Flow chart showing strategy of FISH panel in newly diagnosed and postinduction follow-up AML patients.
|
Cytogenetic abnormality |
Genes involved |
Prognosis |
Conventional karyotyping |
FISH |
Type of FISH probe |
|---|---|---|---|---|---|
|
t(8;21)(q22;q22) |
RUNX1T1, RUNX1 |
Good |
Yes |
Yes |
DC DF |
|
t(15;17)(q24.1;q21) |
PML, RARA |
Good |
Yes |
Yes |
DC DF |
|
t(9;22)(q34.1;q11.2) |
BCR, ABL1 |
Poor |
Yes |
Yes |
DC DF |
|
t(6;9)(p22;q34.1) |
DEK, NUP214 |
Poor |
Yes |
Yes |
DC DF |
|
t(1;22)(p13.3;q13.1) |
RBM15, MRTFA |
Intermediate |
Yes |
Yes |
DC DF |
|
t(?;11)(?;q23.3) |
KMT2A |
Poor |
Yes |
Yes |
BA |
|
inv(16)(p13.1q22)/ t(16;16)(p13.1;q22) |
MYH11, CBFB |
Good |
Yes |
Yes |
BA |
|
inv(3)(q21q26.2)/ t(3;3)(q21;q26.2) |
MECOM |
Poor |
Yes |
Yes |
BA |
|
t(11;?)(p15;?) |
NUP98 |
Adverse |
No |
Yes |
BA |
|
inv(16)(p13.1q24) |
CBFA2T3, GLIS2 |
Adverse |
No |
Yes |
BA/ DC DF |
|
t(7;12)(q36;p13.1) |
MNX1, ETV6 |
Adverse |
No |
Yes |
BA/ DC DF |
|
Monosomy 5/del(5q31/q33) |
CSF1R, EGR1 |
Poor |
Yes |
Yes |
DC/TC del |
|
Monosomy 7/del(7q22/q36) |
KMT2E, CUL1 |
Poor |
Yes |
Yes |
DC/TC del |
|
del(17)(p13) |
TP53 |
Poor |
Yes |
Yes |
DC del |
|
Trisomy 8 |
– |
Intermediate |
Yes |
Yes |
CEP |
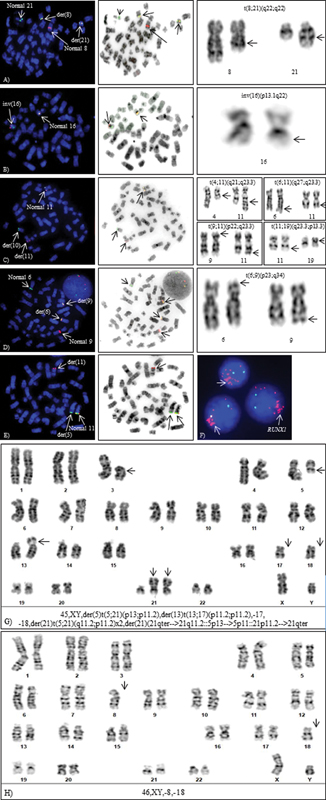
Figure 3:Cytogenetic aberrations detected by FISH and CC. Metaphase-FISH, inverted DAPI and G-banded partial karyotypes (A) t(8;21)(q22;q22), (B) inv(16)(p13.1q22), (C) t(10;11)(p13;q23), (D) t(6;9)(p22;q34.1), (E) cryptic NUP98 rearrangement: t(5;11)(q35;p15.5) by metaphase-FISH, (F) intrachromosomal amplification of RUNX1, (G) Complex karyotype, and (H) Monosomal karyotype.
PML::RARA – t(15;17)(q24.1;q21.2)
PML::RARA fusion is formed due to reciprocal translocation between chromosomes 15 and 17 in APL and is associated with favorable prognosis.[33] Rapid detection of APL by FISH or other techniques is essential due to the high risk of early death and the availability of targeted treatment, ATRA. Unlike the standard t(15;17), complex rearrangements or insertions of the PML and RARA genes result in t(15;17) in 5%-of cases of APL which appear normal by CC. Several variants and cryptic translocations involving RARA have been identified in about 10%-APL cases which include ZBTB16::RARA– t(11;17)(q23;q21), NPM1::RARA – t(5;17)(q35;q21), NUMA::RARA – t(11;17)(q13;q21), STAT5B/RARA – der(17), PRKAR1a::RARA – t(17;17)(q21;q24) or del(17)(q21), BCOR::RARA – t(X:17)(p11;q21), and FIP1L1::RARA –t(4;17)(q12;q21) with differential response to ATRA. FISH with a PML/RARA DC DF and RARA BA probe is useful for rapid detection of PML::RARA or RARA variants leading to prompt therapy.[34] [35] Good prognosis associated with t(15;17) does not appear to be affected by other additional abnormalities like trisomy 8, del(7q), or del(9q).[36]
CBFB::MYH11–inv(16)/t(16;16)(p13.1;q22)
Inv(16)(p13.1q22.1)/ t(16;16)(p13.1;q22.1) leads to fusion of MYH11 at 16p13.1 with CBFB at 16q22.1 and is consistent with favorable prognosis as this aberration is associated with complete remission[37] ([Fig. 3B]). FISH with BA probe is helpful as this rearrangement can be missed by CC in poor morphology metaphases.[38] The most common abnormality along with inv(16) is trisomy 22, trisomy 8, and del(7q).[39]
KMT2A::? – t(11;?)(q23.3;?)
Rearrangements of KMT2A (lysine [K]-specific methyl transferase 2A earlier known as mixed-lineage leukemia 1) are considered poor risk markers. Multiple rearrangements (translocations, insertions, inversions) involving KMT2A gene on 11q23 are found in 3 to 10- of de novo and therapy-related AML. More than 80 translocation partners and 120 reciprocal fusion variants have been documented.[40] Common translocation partner needs to be identified due to variable prognosis, for example, t(9;11)(p22;q23.3) – KMT2A::MLLT3 is considered to have a better prognosis than t(6;11)(q27;q23.3) – KMT2A::MLLT4 and t(10;11)(p12;q23.3) – KMT2A::MLLT10 which predict poor prognosis.[41] Other common translocation partners identified are t(4;11)(q21;q23.3) – KMT2A::MLLT2, t(11;19)(q23.3;p13.3) – KMT2A::MLLT1, t(11;19)(q23.3;p13.1) – KMT2A::ELL [42] ([Fig. 3C]).
BCR::ABL1–t(9;22)(q34.1;q11.2)
Philadelphia chromosome formed due to reciprocal translocation between chromosomes 9 and 22 leads to BCR::ABL1 fusion, is found in 1% AML cases, and is regarded as high-risk marker.[10]
DEK::NUP214–t(6;9)(p23;q34.1)
Fusion of DEK at 6p23 with NUP214 at 9q34.1 results in t(6;9) is frequently associated with basophilia and is seen in both pediatric and adult patients.[43] It can present either as a sole or a part of a complex karyotype and is an adverse risk marker ([Fig. 3D]).
RPN1::MECOM-inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2)
Inversion(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2) involves genes MECOM (EVI1) at 3q26.2 and RPN1 at 3q21.3 contributing as an unfavorable prognostic marker with a poor outcome.[44] Frequently, monosomy 7 accompanies rearrangements of MECOM (3q26.2) in 50%-cases.[45]
RBM15::MRTFA – t(1;22)(p13.3;q13.1)
T(1;22)(p13.3;q13.1) leads to a fusion of RBM15 at 1p13.3 with MRTFA at 22q13.1 and is seen in 3 to 15%-of pediatric AML with median presentation age of 1 to 8 years.[46] [47] This aberration is characteristic of acute megakaryoblastic leukemia with conflicting reports on its prognostic significance, as some series revealed a negative outcome while others suggested a favorable one.[48] [49]
Rearrangements of NUP98
NUP98 rearrangements are frequently found in 5%-of pediatric AML with an unfavorable outcome. More than 30 NUP98 fusion partners have been identified in numerous hematological malignancies.[50] t(5;11)(q35;p15.5) and t(11;12)(p15.5;p13) are cytogenetically cryptic aberrations, found in CN-AML, and cannot be detected by CC, necessitating the use of DC DF FISH probes: NUP98/NSD1 and NUP98/KDM5A, respectively, for the identification of these poor risk abnormalities[51] [52] ([Fig. 3E]).
Monosomy 5/deletion 5q
Deletion of the long arm of chromosome 5 [del(5q)] or monosomy 5 can be reliably detected by CC or FISH using deletion probes. Isolated del(5q) is known to have a favorable outcome in MDS; however, in AML, this aberration is regarded as an adverse prognostic marker associated with complex karyotypes and poor response to intensive chemotherapy with 20 to 30%-of patients achieving complete remission for a short duration.[4] [53]
Monosomy 7/deletion 7q
Monosomy 7 or deletion 7q, seen in approximately 10%-AML cases, is associated as high-risk cytogenetic marker.[6] It frequently occurs in conjunction with other unfavorable cytogenetic abnormalities like complex karyotype (CK), monosomy 5, or deletion 5q, or inv(3).[4]
TP53 deletion/del(17)(p13)
TP53 is a tumour suppressor gene present on short arm of chromosome 17 and its loss is associated with disease progression and dismal outcome. It is found in 3-5%-of adult AML patients. TP53 deletion positive patients have lower WBC counts, are associated with high-risk cytogenetic markers [−5/del(5q), −7/del(7q)], have CKs and poor or no response to standard chemotherapy.[7] [54]
Trisomy 8
Trisomy 8 is seen in approximately 10 to 20%-of AMLs, either as sole or as an additional abnormality with an intermediate prognosis.[55] It is seen as a secondary abnormality with t(8;21), inv(16)/t(16;16), or t(15;17) and does not alter its outcome.
Amplification of Oncogenes
Intrachromosomal amplifications of oncogenes like RUNX1 (21q22) or KMT2A (11q23.3) genes, rarely found in AML, are defined more than three copies on a single chromosome and are associated with poor prognosis and inferior outcomes.[56] [57] These oncogenic amplifications either present as a cluster on a single chromosome (homogeneously stained regions-hsr), as double minutes, or are interspersed throughout the genome.[58] This aberration can be reliably identified by FISH on interphase cells and confirmed on metaphases using LSI RUNX1/RUNX1T1 probe[59] ([Fig. 3F]).
Complex Karyotype
CK is defined as the presence of three chromosomal structural aberrations in the absence of favorable cytogenetic aberrations: t(8;21), inv(16)/t(16;16), and t(15;17)[21] ([Fig. 3G]). AML patients of this subgroup are unresponsive to therapy and have an extremely dismal prognosis.[60] Numerous chromosome abnormalities, such as unbalanced translocations, gains or losses of chromosomal segments, double minutes, oncogenic amplifications, markers, chromothripsis, and ring chromosomes are frequently present in CKs, necessitating the use of both CC and FISH for their detection.[61]
Monosomal Karyotype
Monosomal karyotype features either two autosomal monosomies or one structural abnormality accompanied by one autosomal monosomy ([Fig. 3H]). MK affects up to 20%-of older populations and accounts for about 10%-of all cases of AML and is associated with an unfavorable prognosis with 4-year overall survival of 3%- compared to 13%-in non-MK patients.[62] Recognition of MK, is crucial in order to apply alternative therapy to improve the associated poor prognosis.
Quality Control for Fluorescence In Situ Hybridization Probes
Every laboratory employing FISH testing for diagnostic purposes must establish quality control/quality assurance measures by validating each FISH probe utilized for analysis. Determining cut-off values of every probe is also crucial in the application of FISH for AML and other hematological malignancies as these cytogenetic aberrations are clonal in nature and require extremely high sensitivity and specificity to detect minimal residual disease. Using the inverse beta function, confidence interval around the mean, maximum likelihood, or other statistical methods, every laboratory should uniformly establish the cut-off values for FISH probes. Before putting the probe in use, other factors like probe verification, specificity, and sensitivity should be confirmed and documented for every new lot received.[63]
Conclusion
Although high-resolution molecular profiling methods like single nucleotide polymorphism array, Next generation sequencing and whole-genome sequencing are crucial for discovering new chromosomal abnormalities, especially fusions, they are impractical for routine diagnostic laboratories due to high cost, long turnaround times, and requirement of expertise in bioinformatics. CC will always be the gold standard, preferred method due to its immense utility in genome-wide evaluations. Application of FISH, as an adjunct to CC, is a powerful strategy used today for identifying recurrent genetic abnormalities that offer valuable information for prognosis, risk stratification, and disease diagnosis in hematolymphoid malignancies.
Conflict of Interest
None declared.
Acknowledgment
We thank Mr. Jayraj Kasale, photography section, ACTREC, for his assistance in preparation of figures/images.
Authors' Contributions
H.J. was responsible for concept, design, definition of intellectual content, literature search, and manuscript preparation. D.S. was responsible for manuscript editing and manuscript review. The manuscript has been read and approved by all the authors, and the requirements for authorship have been met and each author believes that the manuscript represents honest work.
Supplementary Material
- Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015; 373 (12) 1136-1152
- Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer 2017; 17 (01) 5-19
- Barrett R, Morash B, Roback D. et al. FISH identifies a KAT6A/CREBBP fusion caused by a cryptic insertional t(8;16) in a case of spontaneously remitting congenital acute myeloid leukemia with a normal karyotype. Pediatr Blood Cancer 2017; 64 (08) DOI: 10.1002/pbc.26450.
- Grimwade D, Hills RK, Moorman AV. et al; National Cancer Research Institute Adult Leukaemia Working Group. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010; 116 (03) 354-365
- Vardiman JW, Thiele J, Arber DA. et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009; 114 (05) 937-951
- Grimwade D, Walker H, Oliver F. et al; The Medical Research Council Adult and Children's Leukaemia Working Parties. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. Blood 1998; 92 (07) 2322-2333
- Byrd JC, Mrózek K, Dodge RK. et al; Cancer and Leukemia Group B (CALGB 8461). Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002; 100 (13) 4325-4336
- Slovak ML, Kopecky KJ, Cassileth PA. et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood 2000; 96 (13) 4075-4083
- O'Donnell MR, Tallman MS, Abboud CN. et al. Acute Myeloid Leukemia, Version 3.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw 2017; 15 (07) 926-957
- Döhner H, Wei AH, Appelbaum FR. et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022; 140 (12) 1345-1377
- Latagliata R, Avvisati G, Lo Coco F. et al. The role of all-trans-retinoic acid (ATRA) treatment in newly-diagnosed acute promyelocytic leukemia patients aged > 60 years. Ann Oncol 1997; 8 (12) 1273-1275
- Ablain J, de The H. Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood 2011; 117 (22) 5795-5802
- Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood 2016; 127 (01) 29-41
- Ramos NR, Mo CC, Karp JE, Hourigan CS. Current approaches in the treatment of relapsed and refractory acute myeloid leukemia. J Clin Med 2015; 4 (04) 665-695
- Mardis ER, Ding L, Dooling DJ. et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009; 361 (11) 1058-1066
- Sholl LM, Longtine J, Kuo FC. Molecular analysis of gene rearrangements and mutations in acute leukemias and myeloid neoplasms. Curr Protoc Hum Genet 2017; 92: 10.4.1-10.4.49
- Bennett JM, Catovsky D, Daniel MT. et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 1976; 33 (04) 451-458
- Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 2011; 117 (19) 5019-5032
- Arber DA, Orazi A, Hasserjian R. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127 (20) 2391-2405
- Khoury JD, Solary E, Abla O. et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia 2022; 36 (07) 1703-1719
- Tallman MS, Wang ES, Altman JK. et al; OCN. Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2019; 17 (06) 721-749
- Mikhail FM, Heerema NA, Rao KW, Burnside RD, Cherry AM, Cooley LD. Section E6.1-6.4 of the ACMG technical standards and guidelines: chromosome studies of neoplastic blood and bone marrow-acquired chromosomal abnormalities. [published correction appears in Genet Med. 2016 Aug;18(8):859] Genet Med 2016; 18 (06) 635-642
- Seabright M. A rapid banding technique for human chromosomes. Lancet 1971; 2 (7731): 971-972
- Jean McGowan-Jordan. Sarah M, Hastings R. An International System for Human Cytogenomic Nomenclature; 2020
- Martens JH, Stunnenberg HG. The molecular signature of oncofusion proteins in acute myeloid leukemia. FEBS Lett 2010; 584 (12) 2662-2669
- Marceau-Renaut A, Duployez N, Ducourneau B. et al. Molecular profiling defines distinct prognostic subgroups in childhood AML: a report from the French ELAM02 Study Group. HemaSphere 2018; 2 (01) e31
- Fröhling S, Kayser S, Mayer C. et al; AML Study Group Ulm. Diagnostic value of fluorescence in situ hybridization for the detection of genomic aberrations in older patients with acute myeloid leukemia. Haematologica 2005; 90 (02) 194-199
- Kearney L. Molecular cytogenetics. Best Pract Res Clin Haematol 2001; 14 (03) 645-669
- Telenius H, Pelmear AH, Tunnacliffe A. et al. Cytogenetic analysis by chromosome painting using DOP-PCR amplified flow-sorted chromosomes. Genes Chromosomes Cancer 1992; 4 (03) 257-263
- Senger G, Lüdecke HJ, Horsthemke B, Claussen U. Microdissection of banded human chromosomes. Hum Genet 1990; 84 (06) 507-511
- Gorczyca W. Cytogenetics, FISH and Molecular Testing in Hematologic Malignancies. 1st ed. Informa UK Ltd.; 2008: 31-108
- Harrison CJ, Hills RK, Moorman AV. et al. Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J Clin Oncol 2010; 28 (16) 2674-2681
- de Thé H, Chomienne C, Lanotte M, Degos L, Dejean A. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 1990; 347 (6293): 558-561
- Sirulnik A, Melnick A, Zelent A, Licht JD. Molecular pathogenesis of acute promyelocytic leukaemia and APL variants. Best Pract Res Clin Haematol 2003; 16 (03) 387-408
- Yin CC, Glassman AB, Lin P. et al. Morphologic, cytogenetic, and molecular abnormalities in therapy-related acute promyelocytic leukemia. Am J Clin Pathol 2005; 123 (06) 840-848
- De Lourdes Chauffaille M, Borri D, Proto-Siqueira R, Moreira ES, Alberto FL. Acute promyelocytic leukemia with t(15;17): frequency of additional clonal chromosome abnormalities and FLT3 mutations. Leuk Lymphoma 2008; 49 (12) 2387-2389
- Shigesada K, van de Sluis B, Liu PP. Mechanism of leukemogenesis by the inv(16) chimeric gene CBFB/PEBP2B-MHY11. Oncogene 2004; 23 (24) 4297-4307
- Hernández JM, González MB, Granada I. et al. Detection of inv(16) and t(16;16) by fluorescence in situ hybridization in acute myeloid leukemia M4Eo. Haematologica 2000; 85 (05) 481-485
- Appelbaum FR, Kopecky KJ, Tallman MS. et al. The clinical spectrum of adult acute myeloid leukaemia associated with core binding factor translocations. Br J Haematol 2006; 135 (02) 165-173
- Ney Garcia DR, De Souza MT, De Figueiredo AF. et al. Molecular characterization of KMT2A fusion partner genes in 13 cases of pediatric leukemia with complex or cryptic karyotypes. Hematol Oncol 2017; 35 (04) 760-768
- Rubnitz JE, Raimondi SC, Tong X. et al. Favorable impact of the t(9;11) in childhood acute myeloid leukemia. J Clin Oncol 2002; 20 (09) 2302-2309
- Schoch C, Schnittger S, Klaus M, Kern W, Hiddemann W, Haferlach T. AML with 11q23/MLL abnormalities as defined by the WHO classification: incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood 2003; 102 (07) 2395-2402
- Oancea C, Rüster B, Henschler R, Puccetti E, Ruthardt M. The t(6;9) associated DEK/CAN fusion protein targets a population of long-term repopulating hematopoietic stem cells for leukemogenic transformation. Leukemia 2010; 24 (11) 1910-1919
- Fonatsch C, Gudat H, Lengfelder E. et al. Correlation of cytogenetic findings with clinical features in 18 patients with inv(3)(q21q26) or t(3;3)(q21;q26). Leukemia 1994; 8 (08) 1318-1326
- Lavallée VP, Gendron P, Lemieux S, D'Angelo G, Hébert J, Sauvageau G. EVI1-rearranged acute myeloid leukemias are characterized by distinct molecular alterations. Blood 2015; 125 (01) 140-143
- Dastugue N, Lafage-Pochitaloff M, Pagès MP. et al; Groupe Français d'Hematologie Cellulaire. Cytogenetic profile of childhood and adult megakaryoblastic leukemia (M7): a study of the Groupe Français de Cytogénétique Hématologique (GFCH). Blood 2002; 100 (02) 618-626
- Oki Y, Kantarjian HM, Zhou X. et al. Adult acute megakaryocytic leukemia: an analysis of 37 patients treated at M.D. Anderson Cancer Center. Blood 2006; 107 (03) 880-884
- Schweitzer J, Zimmermann M, Rasche M. et al. Improved outcome of pediatric patients with acute megakaryoblastic leukemia in the AML-BFM 04 trial. Ann Hematol 2015; 94 (08) 1327-1336
- O'Brien MM, Cao X, Pounds S. et al. Prognostic features in acute megakaryoblastic leukemia in children without Down syndrome: a report from the AML02 multicenter trial and the Children's Oncology Group Study POG 9421. Leukemia 2013; 27 (03) 731-734
- Bisio V, Zampini M, Tregnago C. et al. NUP98-fusion transcripts characterize different biological entities within acute myeloid leukemia: a report from the AIEOP-AML group. Leukemia 2017; 31 (04) 974-977
- de Rooij JD, Hollink IH, Arentsen-Peters ST. et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 2013; 27 (12) 2280-2288
- Thiollier C, Lopez CK, Gerby B. et al. Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J Exp Med 2012; 209 (11) 2017-2031
- Fenaux P, Preudhomme C, Laï JL, Morel P, Beuscart R, Bauters F. Cytogenetics and their prognostic value in de novo acute myeloid leukaemia: a report on 283 cases. Br J Haematol 1989; 73 (01) 61-67
- Seifert H, Mohr B, Thiede C. et al; Study Alliance Leukemia (SAL). The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia 2009; 23 (04) 656-663
- Johansson B, Harrison CJ. Acute myeloid leukemia. In: Heim S, Mitelman F, eds. Cancer Cytogenetics, Chromosomal and Molecular Genetic Aberrations in Tumor Cells. Hoboken, NJ: John Wiley & Sons; 45-139
- Streubel B, Valent P, Lechner K, Fonatsch C. Amplification of the AML1(CBFA2) gene on ring chromosomes in a patient with acute myeloid leukemia and a constitutional ring chromosome 21. Cancer Genet Cytogenet 2001; 124 (01) 42-46
- Tang G, DiNardo C, Zhang L. et al. MLL gene amplification in acute myeloid leukemia and myelodysplastic syndromes is associated with characteristic clinicopathological findings and TP53 gene mutation. Hum Pathol 2015; 46 (01) 65-73
- Albertson DG. Gene amplification in cancer. Trends Genet 2006; 22 (08) 447-455
- Jain H, Shetty D, Roy Moulik N, Narula G, Subramanian PG, Banavali S. A novel case of intrachromosomal amplification and insertion of RUNX1 on derivative chromosome 2 in pediatric AML. Cancer Genet 2021; 254-255: 65-69
- Grimwade D, Walker H, Harrison G. et al; Medical Research Council Adult Leukemia Working Party. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood 2001; 98 (05) 1312-1320
- Mrózek K. Cytogenetic, molecular genetic, and clinical characteristics of acute myeloid leukemia with a complex karyotype. Semin Oncol 2008; 35 (04) 365-377
- Breems DA, Van Putten WL, De Greef GE. et al. Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. J Clin Oncol 2008; 26 (29) 4791-4797
- Shetty D, Talker E, Jain H. Preclinical in-house validation of commercially available fluorescence in situ hybridization probes used in diagnosis of hematological malignancies. Eur J Mol Cancer. 2020; 3 (01) 1-6
Figure 1:Different types of FISH probes. Normal (A) 2R2G, (C) 2F, (E) 2R2G and abnormal (B) 2F1R1G, (D) 1F1R1G, (F) 1R2G, (G) 1R1G, (H) 1G, (I) 3R FISH signal patterns on interphase cells (J) WCP showing t(2;11) and (K) M-FISH showing complex karyotype on metaphase
Figure 2:Flow chart showing strategy of FISH panel in newly diagnosed and postinduction follow-up AML patients.
Figure 3:Cytogenetic aberrations detected by FISH and CC. Metaphase-FISH, inverted DAPI and G-banded partial karyotypes (A) t(8;21)(q22;q22), (B) inv(16)(p13.1q22), (C) t(10;11)(p13;q23), (D) t(6;9)(p22;q34.1), (E) cryptic NUP98 rearrangement: t(5;11)(q35;p15.5) by metaphase-FISH, (F) intrachromosomal amplification of RUNX1, (G) Complex karyotype, and (H) Monosomal karyotype.
- Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015; 373 (12) 1136-1152
- Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer 2017; 17 (01) 5-19
- Barrett R, Morash B, Roback D. et al. FISH identifies a KAT6A/CREBBP fusion caused by a cryptic insertional t(8;16) in a case of spontaneously remitting congenital acute myeloid leukemia with a normal karyotype. Pediatr Blood Cancer 2017; 64 (08) DOI: 10.1002/pbc.26450.
- Grimwade D, Hills RK, Moorman AV. et al; National Cancer Research Institute Adult Leukaemia Working Group. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010; 116 (03) 354-365
- Vardiman JW, Thiele J, Arber DA. et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009; 114 (05) 937-951
- Grimwade D, Walker H, Oliver F. et al; The Medical Research Council Adult and Children's Leukaemia Working Parties. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. Blood 1998; 92 (07) 2322-2333
- Byrd JC, Mrózek K, Dodge RK. et al; Cancer and Leukemia Group B (CALGB 8461). Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002; 100 (13) 4325-4336
- Slovak ML, Kopecky KJ, Cassileth PA. et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood 2000; 96 (13) 4075-4083
- O'Donnell MR, Tallman MS, Abboud CN. et al. Acute Myeloid Leukemia, Version 3.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw 2017; 15 (07) 926-957
- Döhner H, Wei AH, Appelbaum FR. et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022; 140 (12) 1345-1377
- Latagliata R, Avvisati G, Lo Coco F. et al. The role of all-trans-retinoic acid (ATRA) treatment in newly-diagnosed acute promyelocytic leukemia patients aged > 60 years. Ann Oncol 1997; 8 (12) 1273-1275
- Ablain J, de The H. Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood 2011; 117 (22) 5795-5802
- Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood 2016; 127 (01) 29-41
- Ramos NR, Mo CC, Karp JE, Hourigan CS. Current approaches in the treatment of relapsed and refractory acute myeloid leukemia. J Clin Med 2015; 4 (04) 665-695
- Mardis ER, Ding L, Dooling DJ. et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009; 361 (11) 1058-1066
- Sholl LM, Longtine J, Kuo FC. Molecular analysis of gene rearrangements and mutations in acute leukemias and myeloid neoplasms. Curr Protoc Hum Genet 2017; 92: 10.4.1-10.4.49
- Bennett JM, Catovsky D, Daniel MT. et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 1976; 33 (04) 451-458
- Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 2011; 117 (19) 5019-5032
- Arber DA, Orazi A, Hasserjian R. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127 (20) 2391-2405
- Khoury JD, Solary E, Abla O. et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia 2022; 36 (07) 1703-1719
- Tallman MS, Wang ES, Altman JK. et al; OCN. Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2019; 17 (06) 721-749
- Mikhail FM, Heerema NA, Rao KW, Burnside RD, Cherry AM, Cooley LD. Section E6.1-6.4 of the ACMG technical standards and guidelines: chromosome studies of neoplastic blood and bone marrow-acquired chromosomal abnormalities. [published correction appears in Genet Med. 2016 Aug;18(8):859] Genet Med 2016; 18 (06) 635-642
- Seabright M. A rapid banding technique for human chromosomes. Lancet 1971; 2 (7731): 971-972
- Jean McGowan-Jordan. Sarah M, Hastings R. An International System for Human Cytogenomic Nomenclature; 2020
- Martens JH, Stunnenberg HG. The molecular signature of oncofusion proteins in acute myeloid leukemia. FEBS Lett 2010; 584 (12) 2662-2669
- Marceau-Renaut A, Duployez N, Ducourneau B. et al. Molecular profiling defines distinct prognostic subgroups in childhood AML: a report from the French ELAM02 Study Group. HemaSphere 2018; 2 (01) e31
- Fröhling S, Kayser S, Mayer C. et al; AML Study Group Ulm. Diagnostic value of fluorescence in situ hybridization for the detection of genomic aberrations in older patients with acute myeloid leukemia. Haematologica 2005; 90 (02) 194-199
- Kearney L. Molecular cytogenetics. Best Pract Res Clin Haematol 2001; 14 (03) 645-669
- Telenius H, Pelmear AH, Tunnacliffe A. et al. Cytogenetic analysis by chromosome painting using DOP-PCR amplified flow-sorted chromosomes. Genes Chromosomes Cancer 1992; 4 (03) 257-263
- Senger G, Lüdecke HJ, Horsthemke B, Claussen U. Microdissection of banded human chromosomes. Hum Genet 1990; 84 (06) 507-511
- Gorczyca W. Cytogenetics, FISH and Molecular Testing in Hematologic Malignancies. 1st ed. Informa UK Ltd.; 2008: 31-108
- Harrison CJ, Hills RK, Moorman AV. et al. Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J Clin Oncol 2010; 28 (16) 2674-2681
- de Thé H, Chomienne C, Lanotte M, Degos L, Dejean A. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 1990; 347 (6293): 558-561
- Sirulnik A, Melnick A, Zelent A, Licht JD. Molecular pathogenesis of acute promyelocytic leukaemia and APL variants. Best Pract Res Clin Haematol 2003; 16 (03) 387-408
- Yin CC, Glassman AB, Lin P. et al. Morphologic, cytogenetic, and molecular abnormalities in therapy-related acute promyelocytic leukemia. Am J Clin Pathol 2005; 123 (06) 840-848
- De Lourdes Chauffaille M, Borri D, Proto-Siqueira R, Moreira ES, Alberto FL. Acute promyelocytic leukemia with t(15;17): frequency of additional clonal chromosome abnormalities and FLT3 mutations. Leuk Lymphoma 2008; 49 (12) 2387-2389
- Shigesada K, van de Sluis B, Liu PP. Mechanism of leukemogenesis by the inv(16) chimeric gene CBFB/PEBP2B-MHY11. Oncogene 2004; 23 (24) 4297-4307
- Hernández JM, González MB, Granada I. et al. Detection of inv(16) and t(16;16) by fluorescence in situ hybridization in acute myeloid leukemia M4Eo. Haematologica 2000; 85 (05) 481-485
- Appelbaum FR, Kopecky KJ, Tallman MS. et al. The clinical spectrum of adult acute myeloid leukaemia associated with core binding factor translocations. Br J Haematol 2006; 135 (02) 165-173
- Ney Garcia DR, De Souza MT, De Figueiredo AF. et al. Molecular characterization of KMT2A fusion partner genes in 13 cases of pediatric leukemia with complex or cryptic karyotypes. Hematol Oncol 2017; 35 (04) 760-768
- Rubnitz JE, Raimondi SC, Tong X. et al. Favorable impact of the t(9;11) in childhood acute myeloid leukemia. J Clin Oncol 2002; 20 (09) 2302-2309
- Schoch C, Schnittger S, Klaus M, Kern W, Hiddemann W, Haferlach T. AML with 11q23/MLL abnormalities as defined by the WHO classification: incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood 2003; 102 (07) 2395-2402
- Oancea C, Rüster B, Henschler R, Puccetti E, Ruthardt M. The t(6;9) associated DEK/CAN fusion protein targets a population of long-term repopulating hematopoietic stem cells for leukemogenic transformation. Leukemia 2010; 24 (11) 1910-1919
- Fonatsch C, Gudat H, Lengfelder E. et al. Correlation of cytogenetic findings with clinical features in 18 patients with inv(3)(q21q26) or t(3;3)(q21;q26). Leukemia 1994; 8 (08) 1318-1326
- Lavallée VP, Gendron P, Lemieux S, D'Angelo G, Hébert J, Sauvageau G. EVI1-rearranged acute myeloid leukemias are characterized by distinct molecular alterations. Blood 2015; 125 (01) 140-143
- Dastugue N, Lafage-Pochitaloff M, Pagès MP. et al; Groupe Français d'Hematologie Cellulaire. Cytogenetic profile of childhood and adult megakaryoblastic leukemia (M7): a study of the Groupe Français de Cytogénétique Hématologique (GFCH). Blood 2002; 100 (02) 618-626
- Oki Y, Kantarjian HM, Zhou X. et al. Adult acute megakaryocytic leukemia: an analysis of 37 patients treated at M.D. Anderson Cancer Center. Blood 2006; 107 (03) 880-884
- Schweitzer J, Zimmermann M, Rasche M. et al. Improved outcome of pediatric patients with acute megakaryoblastic leukemia in the AML-BFM 04 trial. Ann Hematol 2015; 94 (08) 1327-1336
- O'Brien MM, Cao X, Pounds S. et al. Prognostic features in acute megakaryoblastic leukemia in children without Down syndrome: a report from the AML02 multicenter trial and the Children's Oncology Group Study POG 9421. Leukemia 2013; 27 (03) 731-734
- Bisio V, Zampini M, Tregnago C. et al. NUP98-fusion transcripts characterize different biological entities within acute myeloid leukemia: a report from the AIEOP-AML group. Leukemia 2017; 31 (04) 974-977
- de Rooij JD, Hollink IH, Arentsen-Peters ST. et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 2013; 27 (12) 2280-2288
- Thiollier C, Lopez CK, Gerby B. et al. Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J Exp Med 2012; 209 (11) 2017-2031
- Fenaux P, Preudhomme C, Laï JL, Morel P, Beuscart R, Bauters F. Cytogenetics and their prognostic value in de novo acute myeloid leukaemia: a report on 283 cases. Br J Haematol 1989; 73 (01) 61-67
- Seifert H, Mohr B, Thiede C. et al; Study Alliance Leukemia (SAL). The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia 2009; 23 (04) 656-663
- Johansson B, Harrison CJ. Acute myeloid leukemia. In: Heim S, Mitelman F, eds. Cancer Cytogenetics, Chromosomal and Molecular Genetic Aberrations in Tumor Cells. Hoboken, NJ: John Wiley & Sons; 45-139
- Streubel B, Valent P, Lechner K, Fonatsch C. Amplification of the AML1(CBFA2) gene on ring chromosomes in a patient with acute myeloid leukemia and a constitutional ring chromosome 21. Cancer Genet Cytogenet 2001; 124 (01) 42-46
- Tang G, DiNardo C, Zhang L. et al. MLL gene amplification in acute myeloid leukemia and myelodysplastic syndromes is associated with characteristic clinicopathological findings and TP53 gene mutation. Hum Pathol 2015; 46 (01) 65-73
- Albertson DG. Gene amplification in cancer. Trends Genet 2006; 22 (08) 447-455
- Jain H, Shetty D, Roy Moulik N, Narula G, Subramanian PG, Banavali S. A novel case of intrachromosomal amplification and insertion of RUNX1 on derivative chromosome 2 in pediatric AML. Cancer Genet 2021; 254-255: 65-69
- Grimwade D, Walker H, Harrison G. et al; Medical Research Council Adult Leukemia Working Party. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood 2001; 98 (05) 1312-1320
- Mrózek K. Cytogenetic, molecular genetic, and clinical characteristics of acute myeloid leukemia with a complex karyotype. Semin Oncol 2008; 35 (04) 365-377
- Breems DA, Van Putten WL, De Greef GE. et al. Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. J Clin Oncol 2008; 26 (29) 4791-4797
- Shetty D, Talker E, Jain H. Preclinical in-house validation of commercially available fluorescence in situ hybridization probes used in diagnosis of hematological malignancies. Eur J Mol Cancer. 2020; 3 (01) 1-6